Alignment program evaluation
The performance of three different aligners, BWA-MEM, Bowtie2, and SOAP2, was evaluated using Illumina paired-end read datasets from 52 domesticated tomato, 30 related wild relatives (Supplemental Table 1) [35], and simulated genomic sequences from different crops. Mapping percentage, alignment accuracy, and processing time for each aligner were evaluated.
The ability to align reads to a domesticated tomato reference genome, Solanum lycopersicum [36], was assessed using default and tuned parameters on Bowtie2 (Bowtie2 and Bowtie2-tuned), SOAP2 (SOAP2 and SOAP2-tuned), and default parameters for BWA-MEM. Parameter tuning (see details in Methods) for Bowtie2 and SOAP2 was necessary to attempt to match the mapping percentage to the default used by BWA-MEM. BWA-MEM showed the highest alignment percentage, 99.54% and 95.95% in domesticated and wild relatives, respectively, while SOAP2 showed the lowest alignment percentage, 91.25% and 40.58%, respectively (Supplemental Table 2). In the domesticated tomato datasets, all of the five alignment settings resulted in more than 90% mapping percentage with standard deviation ranging from 0.34% to 3.77% (Figure 1A). Greater variation in mapping percentage existed when analyzing the sequences from wild species with standard deviation ranging from 1.91% to 24.25%. The mapping percentage in the wild tomato samples displayed a bimodal distribution (Figure 1A). The distribution of the group with higher alignment percentage contained wild species that were closely related to domesticated tomatoes, whereas the lower group contained distantly related wild species based on previous domestication and diversity studies [3, 37]. Alignment percentage was found to be negatively correlated with the IBS distance of each sample to the S. lycopersicum reference genome (Figure 1B). When the sample was distantly related to the reference genome, BWA-MEM resulted in the highest mapping percentage and SOAP2 resulted in the lowest mapping percentage. In terms of processing time, SOAP2 was the fastest aligner in both domesticated and wild tomato datasets, and it was up to five times faster than the slowest alignment setting, Bowtie2-tuned (Supplemental Figure 1A).
We next determined whether greater alignment percentage or shorter alignment time could result in tradeoffs on accuracy and sensitivity by using simulated datasets and calculating the ratio of true positive (TP), false positive (FP) and false negative (FN) alignments. Simulated datasets were derived from the reference genome by permuting fragment sizes, and number of SNPs or size of small indels per read. For all alignment methods, the ratio of FP alignment increased as more SNPs or indels were introduced per read (Figure 1C-D) when the fragment size was fixed at 600 nt. When the number of introduced SNPs was equal or less than 2, the average percent of FP alignments BWA-MEM, Bowtie2-tuned and SOAP2-tuned was 0.94%, 1.15% and 0.88%, respectively (Figure 1C). When the number of introduced SNPs was greater or equal to 4, the average FP alignment rate of BWA-MEM, Bowtie2-tuned increased to 6.41% and 2.54%, respectively, while SOAP2, and SOAP2-tuned were no longer able to find alignments. BWA-MEM was the only aligner that was capable of finding TP alignments with 15 SNPs per read with FP alignment rate of 18.26%. Similar results were also observed in the indel simulation experiment (Figure 1D). Only BWA-MEM was able to find TP alignments of reads with INDELs up to 40 nt in size at the cost of 26% false alignments.
To indirectly determine the true vs false positive rates of BWA-MEM and Bowtie2 in real data, one million randomly selected reads from six samples (2 S. lycopersicum, 2 S. pennellii and 2 other wild relatives) were aligned to both S. lycopersicum and Solanum pennellii reference genomes [38]. The positions of alignments with mapping quality (MQ) ³40 were compared against the synteny map of the genome generated by nucmer [39]. When the alignment position of read matched to the nucmer conversion of the S. lycopersicum coordinate to the S. pennellii coordinate, the read was considered to be syntenic. If the positions did not match, the read was considered non-syntenic. BWA-MEM was able to align approximately 4.22 times more reads per sample than Bowtie2 (Supplemental Table 3), but only 65.71% (SD ± 2.68%) of these alignments were considered as syntenic compared to 88.17% (SD ± 1.59%) of Bowtie2 alignments.
To extend the study to other crop species, simulated sequencing datasets were generated from rice, soybean, maize and wheat reference genomes by varying the mutation rate from 0.001 to 0.2 (Figure 1E). In these studies, both SNP and INDEL were included in the simulation. When the mutation rate is equal to or lower than 0.04, BWA-MEM was able to align at least 92% of the sequences correctly for rice, tomato and soybean, whereas it was only able to correctly align 81.5% and 82% of the sequences for maize and wheat, respectively. As mutation rate increased, difference in both true positive and false positive alignment was seen among different crops. On average, BWA-MEM was able to find 18.1%, 20.2% and 17.0% more true positive alignments in rice, tomato and soybean than in wheat and maize at mutation rate 0.08, 0.1, and 0.15, respectively. On the other hand, BWA-MEM was able to generate 18.8%, 22.5%, and 24.5% less false positive alignments in rice, tomato and soybean than in wheat and maize at mutation rate 0.08, 0.1, and 0.15, respectively.
Variant Calling Program Comparison
Four variant datasets were produced from the permutation of the aligners, Bowtie2-tuned, and BWA-MEM, and the variant callers SAMtools-mpileup and GATK-HC using 52 domesticated and 30 wild tomatoes. Results showed nearly a two-fold difference in the number of unfiltered SNPs ranging from 69.2M to 133.7M. A greater difference in the variant count in wild species was observed than that found in domesticated ones (Table 1). In domesticated species, dataset sizes ranged from 11.8M to 17.8M unfiltered SNPs, while in wild species it ranged from 66.4M to 128.3M. The primary determinant of variant count between datasets was whether Bowtie-2 or BWA-MEM was used.
To further evaluate the differences in the ability of identifying variants, both individual-level and population-level simulated datasets were generated with varied mutation rate, sequencing coverage and population size. In the simulated population-level datasets, evaluation was performed on both raw and filtered variants. In the comparison of raw variants, GATK-HC was able to identify more true SNPs at the cost of accuracy as sequencing coverage increased in diversity populations. At 5x and 10x coverages, SAMtools-mpileup was able to identify similar recall ratio with higher precision ratio than GATK in the low diversity population. When dealing with high diversity populations, GATK-HC always outperformed SAMtools-mpileup in both precision and recall aspects (Supplemental Figure 2A). In the comparison of raw INDELs, GATK-HC always outperformed SAMtools-mpileup in terms of precision and recall in the low diversity population. In the high diversity populations, GATK-HC was able to identify greater number of true INDELs at the cost of accuracy (Supplemental Figure 2B).
In the filtered SNP results, when the sequencing coverage is at 5x and 10x, GATK-HC was able to result higher precision ratio in all coverage and diversity permutations without compensating the recall ratio (Figure 2A). In the 1x coverage simulation dataset, even though SAMtools-mpileup identified variants with lower precision ratio, it was able to result higher recall ratio in the dataset. In the filtered INDEL results, GATK-HC always outperformed SAMtools-mpileup in terms of precision and recall ratio in the low diversity population. In the high diversity population, SAMtools-mpileup resulted higher precision ratio at the cost of much lower recall ratio (Figure 2B). Noticeably, SAMtools-mpileup was only able to result 3.08% and 1.61% recall ratio in the high diversity populations for SNPs and INDELs, respectively.
In the individual-level simulated dataset, a consistent pattern of trade-off between precision and recall was observed. SAMtools-mpileup was able to generate higher precision ratio for both SNPs and INDELs, however, GATK-HC was able to result higher recall ratio for both SNPs and INDELs as coverage and mutation rate increased in most case (Supplemental Figure 3A-D). Among four different crop species, rice, tomato and soybean has similar results in both variant calling programs. Nevertheless, results from simulated maize datasets had lower precision and recall ratios. Noticeably, when the mutation rate is at 0.1 and 0.15, both variant calling programs resulted lower precision ratio for SNP detection as coverage increased. Maize datasets had the largest magnitude of reduction in precision whereas other crop species resulted similar reduction.
Wild reference genome alignment and variant calling
The large increase in the number of SNPs in wild samples was expected due to both greater distance from the domesticated reference genome and increased diversity relative to the domesticated samples. Expectedly, as the distance from the reference genome increased, a greater proportion of reads was unmapped. The variants in these unmapped reads, especially in the wild species, could represent “missing diversity”. To test this hypothesis, we evaluated how variants discovery in these 82 tomato samples were changed by using a wild reference genome (S. pennellii) [38].
The read alignment to the S. pennellii reference was performed under identical settings as above. As previously seen, BWA-MEM showed the highest mapping percentage and SOAP2 showed the lowest (Figure 3A). In general, mapping percentage in domesticated and wild tomato groups were similar regardless of aligner settings used (Figure 3A). The single outlier with high alignment percentage was a S. pennellii sample with an alignment of 95.13% (or 99.69%) as opposed to 34.22% (or 94.87%) against the S. lycopersicum reference using SOAP2-tuned (or BWA-MEM). Concurrent with these results, the 82 samples, except the S. pennellii sample, had similar IBS distances to the reference genome. As with the S. lycopersicum reference, alignment percentage to the S. pennellii reference was inversely proportional to IBS distance to the reference genome (Figure 3B), suggesting this relationship was independent of reference genome used.
To investigate how diversity estimation varied by reference genome, reads from randomly selected eight domesticated tomatoes and eight wild relative accessions were aligned to the S. pennellii reference. Alignment to the S. pennellii reference genome generated a total of 96,712,749 unfiltered SNPs and 59,944,499 filtered SNPs, while a total of 77,718,102 raw SNPs and 53,036,666 filtered SNPs were identified using the S. lycopersicum reference genome. Compared to using the S. lycopersicum reference genome, significantly more SNPs were identified from 8 domesticated tomato samples when S. pennellii reference genome was used for variant discovery (Supplemental Figure 4A).
To further investigate the source of this additional variation, a cross-reference comparison was performed between SNPs identified using S. pennellii and S. lycopersicum reference genomes. One hundred nucleotides of DNA sequence flanking each filtered SNP identified using one reference genome was aligned to the other reference genome. Results in the Figure 3C showed that majority of the filtered SNPs identified in the S. pennellii located on the synteny path of S. lycopersicum genome. In the S. lycopersicum sample, and similarly, majority filtered SNPs identified using S. pennellii reference were located on the synteny path of S. lycopersicum genome. This result indicated that using S. pennellii reference genome, we were able to identify SNPs that were fixed in the S. lycopersicum domesticated varieties.
Since these SNPs were fixed in S. lycopersicum, they would not have been identified from alignment to the S. lycopersicum reference. Outside of these fixed SNPs in the domesticated species, 4.55% of flanking sequences of SNPs identified using S. pennellii genome in chromosome 1 could not be mapped to the S. lycopersicum reference. Similarly, 11.15 % of the flanking sequences of SNPs identified in the S. pennellii sample using the S. pennellii genome were not found in the S. lycopersicum genome (Figure 3C). Switching to the domesticated reference genome, 7.13% of the downstream sequences of SNPs identified in a S. lycopersicum sample using S. lycopersicum genome could not be found in the S. pennellii genome (Supplemental Figure 4B). These results indicated that a great portion of variation in the wild species would be missed if a single domesticated genome was used as the reference, and vice versa.
Hard-filtering and machine-learning based variant filtering
Variant filtering is required to minimize both false positive and negative genotype calls. Comparisons were made between three variant filtering methods: setting empirical hard-cutoffs (HARD) on metrics such as read depth, strand bias, and variant quality and so on, a newly implemented machine-learning based (ML) variant filtering [14], and a combination between HARD and ML (COMBINED) filtering. Filtered datasets generated from the 602 WGS tomato datasets, including a wide range of domesticated and wild tomato samples [25], were analyzed. A training dataset of 8401 markers from SolCap was used for the training phase of ML [40]. The SolCap is a high confidence dataset consisting of verified markers previously used in genetic studies. In the COMBINED method, the HARD filters were first applied to SolCap to remove low-confidence markers and yield a training set of 7,633 variants. Results indicated that the HARD-filtered method retained the fewest SNPs (94.2M), which was 26.3% and 7.1% fewer than ML-filtered (127.8M) and COMBINED-filtered (101.4M) datasets, respectively (Supplemental Table 4). SNPs in the first 10 million bases in Chromosome 1 (Supplemental Table 4) were cross-compared between the three datasets. 70% of SNPs in this segment were shared among all three filtered datasets (Supplemental Figure 5A), while each dataset had a subset of unique variants.
Two methods were used to indirectly infer the quality of filtered datasets: recapitulation of diversity estimates generated by a “gold standard” set of 22,336,965 SNPs (See details in Methods) in the form of PCA (Supplemental Figure 5B) and IBS analyses (Supplemental Figure 5C), and calculation of LD decay distance for each filtered dataset. SNPs identified by all three filtering methods were removed for this analysis so that the efficacy of each method could be evaluated independently. The underlying assumption of these analyses is that true diversity would recapitulate the known population structure, whereas the population structure would begin to break down as the number of artifacts increased. Using the “gold standard” variant dataset, samples were grouped into four clusters based on PCA and IBS results. All three filtering methods were able to resolve Cluster 1 and Cluster 4, whereas the HARD and ML filtering methods were not able to clearly resolve Cluster 2 from Cluster 3 (Figure 4A-B). In contrast, the COMBINED filtering method was able to identify all four original clusters to reconstruct the population structure of 82 Solanum genomes (Figure 4C).
Next, the contribution of false positive SNPs in each filtered dataset was evaluated by calculating the rate of LD decay. The assumption was that false positive SNPs were random noise that would be found not in LD with nearby SNPs. Therefore, the apparent rate of LD decay in a dataset would increase as the number of false positives increased. As predicted, a greater rate of LD decay was found in all three filtered datasets than that found in the high-confidence dataset. Of the three filtered datasets, the COMBINED method, however, had the lowest rate of LD decay (Figure 4D) approximating the rate of LD decay seen in the high-confidence SNP dataset.
To quantitively measure the difference between hard filtering and machine-learning based filtering, simulated datasets with varied population size, mutation rate and sequencing coverage were generated (Supplemental Figure 6A-B). In the low diversity population datasets, machine-learning based SNP filtering always outperformed hard SNP filtering by 7.38% and 14.14% on average for precision and recall ratio, respectively. In terms of INDEL filtering in the low diversity dataset, machine learning based filtering and hard filtering resulted comparable precision results, however, machine learning based filtering was able to result 12.49% higher recall ratio than hard filtering. In the high diversity population, SNP and INDEL had similar results from different filtering methods. Minor difference was observed in the recall ratio between machine-learning based and hard filtering. No difference was found in the precision ratio between machine-learning based and hard filtering in the high diversity population.
Two-step imputation method
Missing genotypes, possibly due technical limitations, are commonly resolved via imputation. In human studies, standard imputation methods leverage linkage disequilibrium (LD) and reference panels [41]. Beagle 4.1 is a commonly used imputation algorithm in plant studies that can function with or without a reference panel. To determine the importance of a reference panel for SNP imputation, both LD-based and reference panel-assisted imputation were applied to several datasets. A reference panel of 22,336,965 high-confidence, phased SNPs was generated from 82 high coverage (30x) WGS tomato datasets. Imputation results were compared between the two methods. In the first method, missing SNPs were imputed without a reference panel. In the second method, imputation was performed in two steps: in the first step a reference panel was used to impute missing calls only for missing reference variants; and then a second step was employed to impute the remaining missing, non-reference SNPs. Samples were placed in four groups and varying percentages of high confidence genotypes were masked to act as “missing” data (See details in Methods). The concordance (r-squared) between the original masked and imputed genotypes was calculated to estimate imputation accuracy.
Results showed that no difference between LD-based and 2-step imputation was observed in 100 domesticated (DOM) tomato samples (Figure 5A) or the 50 Solanum pimpinellifolium (PIM) samples (Supplemental Figure 7A) datasets. In the dataset of 200 randomly selected tomato samples (RANDOM), at 47% missing data, a 4% difference was observed (Supplemental Figure 7B). When the parameter of missing percentage was set at 72%, 2-step imputation methods showed 60% higher accuracy than LD-based imputation in the dataset of 36 wild tomato species (WILD) (Figure 5B). High LD between SNPs may reduce the need for a reference panel in imputation. The calculated LD decay for each dataset showed that DOM had the slowest LD decay and WILD had the fastest LD decay (Supplemental Figure 7C). Due to the fact that limited samples of wild tomato were available, the number of samples we used in the simulation in DOM (100) was also considerably higher than that in WILD (36). As such, considerably more information was present in the DOM dataset for imputation, as opposed to the WILD dataset which not only had a smaller number of samples but also contained multiple species. To determine if LD continued to be sufficient for imputation in small domesticated panels when the amount of missing data was considerable, 15 randomly selected domesticated tomato samples that were also included in the reference panel (15-DOM-REF) had up to 85% of their genotypes masked. Both methods were applied to the 15-DOM-REF dataset. The results showed two-step imputation was 9.25 times more accurate than Beagle v4.1 direct imputation by when the missing percentage was 85% (Figure 5C).
{kind=link}
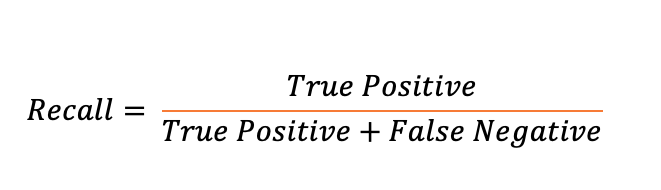{kind=link}