The present study describes mutations that induce constitutive activation or impaired signal transduction in the eelLHR consistent with previously reported mammalian LHR mutations that cause FMPP and elevated cAMP levels in the absence of agonist. Thus, we constructed eelLHR mutants containing single point mutations in five distinct amino acid residues that were highly conserved among GPCRs. These mutations have been shown to stimulate basal cAMP responsiveness or attenuate agonist-induced activation in a dose-dependent [12, 24–27].
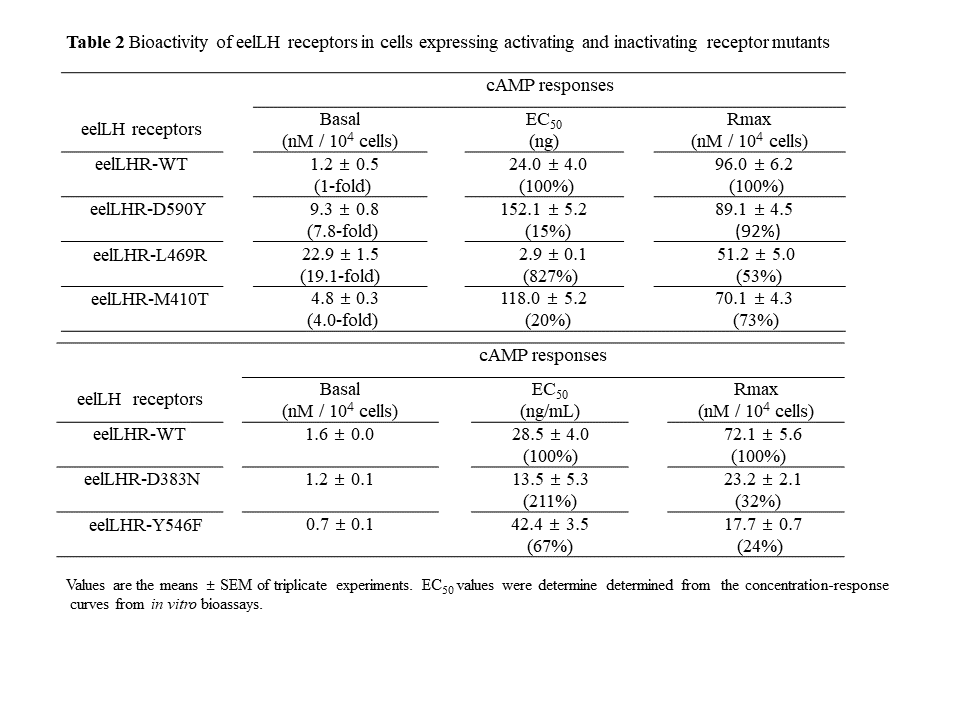
Many studies have suggested that the similar dynamic modification of mammalian LHRs is closely involved in the activity of G proteins [5, 12, 25]. In humans, mutations have previously been described that constitutively activate hLHR and cause FMPP [13, 19]. The differences observed in the phenotypic appearance of FMPP may be accounted for by distinctions in basal hLHR activity [13]. In a previously studied case from Scotland, a patient exhibited signs of pubertal development at 1 year of age [28]. This case was found to have the D578Y mutation (equivalent to D590Y in the eelLHR), which is the mutant that induces higher basal cAMP production than that of the eelLHR-WT. The residues Met-410, Leu-469, Asp-590, Asp-383, and Tyr-546 in eelLHR are conserved among LHRs, suggesting that these residues are important for normal receptor function [24, 25]. Receptor mutations in these residues in the eelLHR have not previously been described.
In the present study, we showed that the three activating mutations in eelLHR resulted in a distinctly increased cAMP response under basal conditions, suggesting that these mutations cause constitutive activation of the eelLHR, as also seen in the FMPP-causing mutations in hLHR. Compared to the eelLHR-WT, the eelLHR-M410T, -L469R, and -D590Y mutants produced a 4-, 19.1-, and 7.8-fold increase, respectively, in the basal cAMP response in CHO-K1 cells, indicating that these three mutants were continuously active, as previously reported in mammalian LHRs [17, 19, 25]. Cells expressing the hLHR-M398T mutant exhibited high basal cAMP levels [7]. The same mutation has been detected in an FMPP patient and in the patient's mother and brother [17]. This mutation is of special interest, one member of this family has the mutation, but with no evidence of precocious puberty [18]. The basal levels of cAMP production were 15- to 25-fold higher in the M398T mutant receptor compared to the WT receptor [5]. In this paper, we also described a constitutively activating mutation in the eelLHR, M410T, which is located in the same second transmembrane region. The L457R mutation (equivalent to L469R in eelLHR) was the first activating mutation identified in hLHR and cells expressing this mutant receptor exhibit markedly higher basal cAMP levels (7-to 14-fold) than that of the WT receptor [19]. We have also reported that basal cAMP responses in cells expressing rLHR-L435R (equivalent to L469R in eelLHR) display a 47-fold increase in the absence of agonist and do not react to hCG with a further stimulation of the cAMP response [25]. In the hLHR, the complex of hLHR-L457R and hCG does not migrate to the lysosomes, but most of it is returned to the cell surface and hormone degradation is hardly detectible [29]. These results are consistent with our current data, showing that the L469R mutant of the eelLHR remarkably increased (19.1-fold) the basal cAMP response in the absence of agonist. However, the maximal cAMP response to agonist was approximately 53% lower than that of the WT receptor, as previously described in hLHR [25]. Thus, our data suggested that the constitutively active mutant, L469R, was easily distinguishable from agonist-activated eelLHRs analyzed in this study.
The aspartic acid residue at position 590 is conserved in all LH receptors, including the eelLHR, but is not found in any other GPCRs. The D578Y mutant (equivalent to D590Y in the eelLHR) was first reported to be inherited in an autosomal dominant and is associated with signs of puberty by 4 years of age. This mutant hLHR results in a 4.5-fold increase in the cAMP response under basal conditions, with an EC50 similar to that of the WT receptor. Agonist-independent stimulation of cAMP production by this mutant receptor represented 42% of the maximal stimulation [6]. This is consistent with our results showing that the eelLHR-D590Y mutation, at a conserved in glycoprotein hormone receptor site, resulted in constitutive receptor activity.
Kosugi et al. [12] suggested that the Asp578 side chain in hLHR has the most appropriate position to act as a hydrogen bond acceptor and is significant for stabilizing the impaired state of the LHR. Other constitutively activating mutations (Ile542Leu, Asp564Gly, Met571Ile, and Cys581Arg) have been identified by analyzing genomic DNA from 32 unrelated FMPP [13]. These sites are preserved among glycoprotein hormone receptors, suggesting an important function in the receptor signaling pathway. These data suggest that the specific nucleotide regions, 1624–1741 in hLHR, are an important point for heterogeneous occurrence mutations that activate the receptor and cause FMPP. The basal cAMP production of activating rat LHR mutant, rLHR-D556Y, also exhibits a 25-fold increase in the absence of agonist, but responds to agonist with a normal increase in cAMP stimulation [25]. Based on the activation and the results summarized above, we expected that eelLHR mutations that induce continuous activation would result in specific changes to the receptor-ligand complex. The D590Y mutant induced an elevated basal cAMP level corresponding to approximately 10% of the maximal cAMP response. However the L469R mutant induced a highly elevated basal cAMP level corresponding to approximately 44% of the maximal cAMP response. These results suggested that the configurations of these three mutants induced different signal transduction pathways, resulting in different maximal cAMP responses to LH.
In the inactive mutants, the highly conserved amino acids present in the second TM helix (codon 383) and in the fifth TM helix (codon 546) were mutated to asparagine and phenylalanine, respectively. As predicted from results obtained with other GPCRs [24–27], these mutations (eelLHR-D383N and eelLHR-Y546F) were expected to impair signal transduction. Cells expressing rLHR-D383N display a rightward shift in the EC50 for cAMP stimulation, but normal maximal levels [24]. The Y524F mutant in rLHR was also a signaling-impairing mutation. Cells expressing this mutant exhibit normal cAMP levels in the absence of agonist, however, their maximal response to agonist is only 14% compared to the WT receptor [25]. These results are consistent with our data, showing that D383N and Y546F are signaling-impairing mutations in the eelLHR. The maximal response of these mutant receptors to agonist was only 24–32% of the maximal response of eelLHR-WT. The internalization of the inactivation mutants, rLHR-D383N and rLHR-Y524F, was much slower than the rLHR-WT [24]. In the present study, D383N and Y546F mutations were predicted to induce the inactivation of the eelLHR. However, these mutations did not completely impair signal transduction in the eelLHR. Thus, we suggest that these mutations are system-dependent or species-specific and therefore, may not have the same effects in fish systems.
{kind=link}
{kind=link}