We identified long-term clinical and laboratory findings and bile acid profiles in 7 Japanese patients with BASD including 5 patients treated with CDCA and 1 with UDCA; the other patient underwent liver transplantation. All patients had improved liver dysfunction, freedom from treatment complications, normal growth and development, and absence of psychosocial problems. Accordingly, the long-term outcomes of these Japanese patients with BASD has been good.
In Europe and the US, 3β-HSD deficiency is about 8 times more common than 5β-reductase deficiency.[15-17] In Saudi Arabia, 2.7% of patients with cholestasis in infancy were found to have BASD.[18] In Japan, our bile acid analyses in 1010 infants with cholestasis lacking obvious cause between 1996 and 2017 showed a BASD prevalence of 0.7%, with equal numbers of cases for 3β-HSD deficiency and 5β-reductase deficiency. Our experience as well as previous reports from East Asia suggests that 3β-HSD deficiency is less prevalent in East Asia than in Europe or the US.[8-11, 13, 19, 20] On the other hand, oxysterol 7α deficiency may be more prevalent in East Asia.[11, 21-23]
CA (10 to 15 mg/kg/day) has been approved in Europe and the US for treating patients with 3β-HSD deficiency and 5β-reductase deficiency, making it the primary bile acid therapy for these BASD in most of the West.[14-17] In particular, CA has a very good therapeutic effect in 3β-HSD deficiency. However, CA monotherapy is insufficient for 5β-reductase deficiency.[18] UDCA treatment is used initially until a diagnosis of 5β-reductase deficiency is made. In early infancy, 5β-reductase deficiency may be exacerbated by CA. In some instances, liver failure may ensue even after CA treatment is started. In consideration of reported mortality, CDCA typically is used in combination with CA to increase the therapeutic effect.[17, 18, 24] In Japan, CDCA is used instead of CA for treatment of BASD, since the latter is not available for clinical use in Japan; the same is true for China.[25] We have obtained a reliable therapeutic effect from CDCA, and believe that a low dose of CDCA (5 to 10 mg/kg/day) can obtain sufficient benefit.[10, 13] Low doses are particularly important because CDCA is more hepatotoxic than CA. CDCA as a treatment for 3β-HSD deficiency and 5β-reductase deficiency has been described in previous reports.[24, 26] We consider low-dose CDCA to be effective in both 3β-HSD deficiency and 5β-reductase deficiency making this treatment particularly important where CA is not available. If CA obtains approval in Japan, we would like to use it in 3β-HSD deficiency and 5β-reductase deficiency, but we believe that patients in East Asia might require only a low dose of CA (5 to 10 mg/kg/day) as opposed to the doses up to 15 mg/kg/day that often are given in Europe and the US.
Before CDCA treatment in our 3 patients with 3β-HSD deficiency, unusual bile acids accounted for 87.5% to 100% and 94% to100% of serum and urinary bile acids, respectively; for our 2 patients with 5β-reductase deficiency, these respective percentages were 54.7% to 91.6% and 98.4% to 99.7% (Tables 2 and 3). During treatment, percentages of unusual bile acids among serum TBA decreased except in Patient 5; in urine, however, these percentages remained high (Tables 2 and 3). However, the concentration of unusual bile acids in urine clearly decreased. In our study, details of bile acid dynamics in hepatocytes are not known, but liver function in our patients improved. This suggests that CDCA treatment suppressed synthesis of unusual bile acids, while the remaining unusual bile acids were excreted via the kidneys. During CDCA treatment, the decreasing concentrations of unusual bile acids in serum and urine resulted from a negative feedback effect by CDCA upon hepatocytic CYP7A1. Concentrations of unusual bile acids in serum decreased except in Patient 5, and hepatic function improved. Given the renal excretion, evaluation of unusual bile acids during CDCA treatment requires bile acid analysis in serum and urine by GC-MS rather than simply determining the percentage of unusual bile acids among urinary TBA. Both concentrations and percentages of unusual bile acids in serum obtained by this analytic method are necessary.
Bile acid analysis showed disappearance of unusual bile acids immediately after liver transplantation in our patient with oxysterol 7α deficiency (Table 4). Recent reports described that oxysterol 7α deficiency can be treated with CDCA treatment.[23, 27] Since liver function rapidly deteriorates in oxysterol 7α deficiency, early diagnosis and initiation of CDCA therapy are particularly urgent.
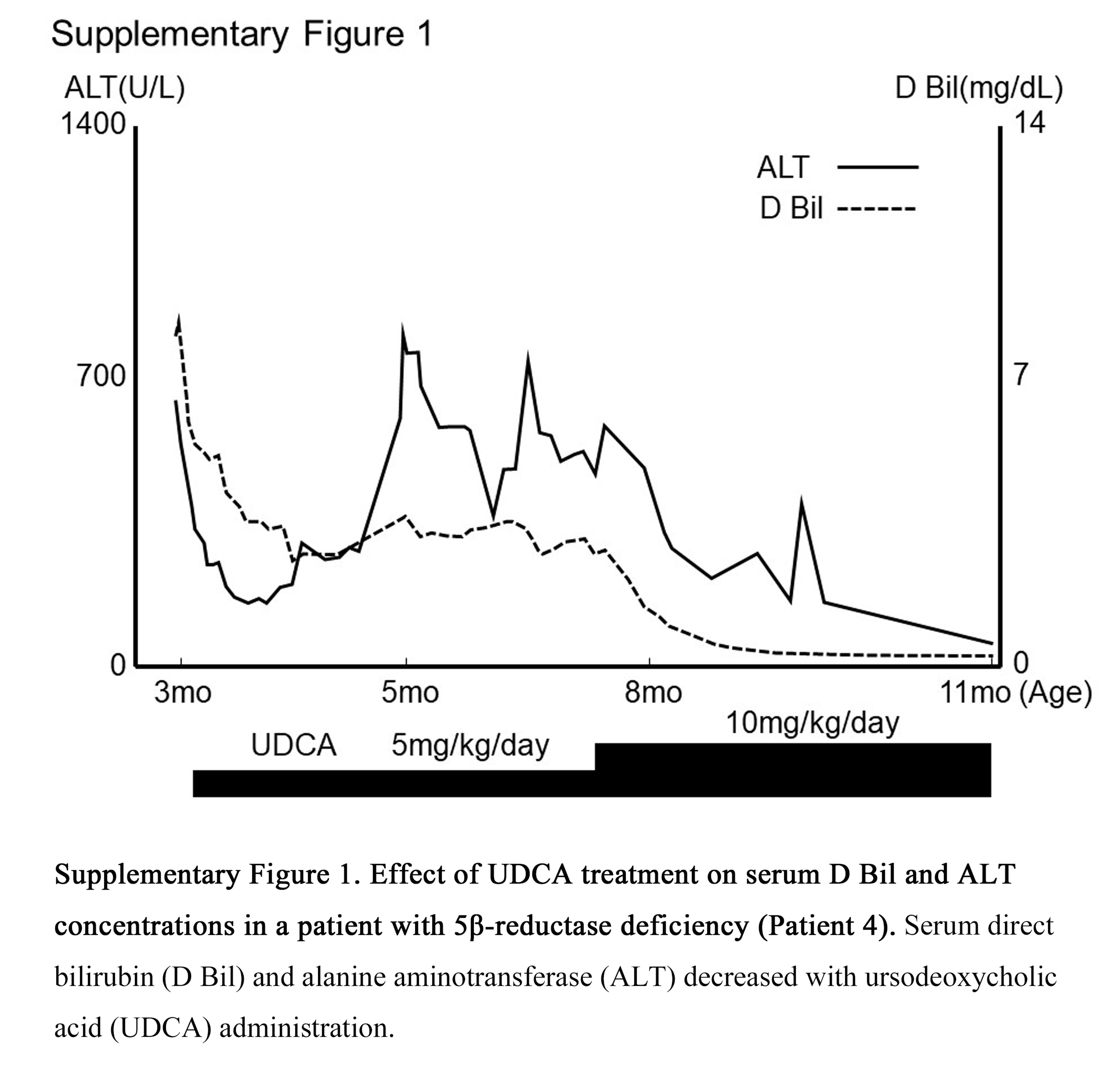
The somewhat enigmatic Patient 4 in Table 5 was diagnosed with 5β-reductase deficiency based on bile acid profiles and genetic analysis. Based on parental wishes, she was treated only with UDCA, which was discontinued at 12 months of age when her liver function had fully recovered and she was free of symptoms. With no treatment, this healthy state has continued through the most recent follow-up at 16 years. We speculate that her favorable course might be related to the small amounts of allo-bile acids, such as allo-cholic acid and allo-chenodeoxycholic acid, detected in serum and urine (Table 5). Allo-bile acids presumably are synthesized from 3-oxo-Δ4-bile acids, which normally would be transformed to 5β-bile acids by 5β-reductase. Fortunately, the toxicity of allo-cholic acid is only on a par with that of cholic acid.[28] Allo-bile acids also are efficiently excreted into the bile via transport systems apart from the bile salt export pump, such as multidrug resistance protein 2 (MRP2).[28, 29] This would account for the very limited quantities of allo-bile acids in the patient’s serum and urine. For this reason, fat-soluble vitamin deficiency and other symptoms have not occurred since discontinuation of treatment. In general, however, allo-bile acids in excess of 3-oxo-Δ4-bile acids can cause significant liver dysfunction.
Unlike allo-bile acids, ketonic bile acids (3-oxo-Δ4-bile acids) are highly toxic to hepatocytes and difficult to excrete into bile.[2] Many ketonic bile acids are discharged into the blood via MRP3 on the basolateral membrane of hepatocytes, followed by renal excretion.[30] Larger amounts of ketonic bile acids in urine than in serum therefore are found. This excretion mechanism increases in activity during late infancy. These findings resemble those that we have seen in bile acid analyses at the beginning of primary bile acid (CDCA) treatment for BASD such as 5β-reductase deficiency. However, at this time the proportions of therapeutically administered bile acids, such as cholic acid and/or chenodeoxycholic acid, increase. Large amounts of 3-oxo-Δ4-bile acids therefore found in serum and urine in neonates and younger infants with 5β-reductase deficiency, while later in infancy 3-oxo-Δ4-bile acids gradually decrease in serum but are abundant in urine. In neonates and young infants with 5β-reductase deficiency, cholestatic liver dysfunction becomes evident, and primary bile acid treatment is necessary. On the other hand, primary bile acid treatment sometimes may not be required after 1 year of age. Allo-bile acids are excreted into bile via MRP2. Ketonic bile acids are transported into the blood via MRP3 and then excreted by the kidneys. Activity of MRP3 increases during infancy, equaling that in an adult at about 1 year. At that point, if ketonic bile acids are excreted adequately, liver function improves and primary bile acid treatment might be no longer be necessary. In another patient with 5β-reductase deficiency (Patient 5 in Table 3), discontinuation of primary bile acid treatment for 1 month did not adversely affect liver function.[10]
In summary, we believe that 3β-HSD deficiency, which is not common in East Asia, should be adequately treatable with CA given that disease progression is slow. In contrast, 5β-reductase deficiency and oxysterol 7α deficiency rapidly progress to liver cirrhosis or cholestatic liver failure in infancy. We therefore believe that in infancy, prompt treatment with CDCA or a combination of CA and CDCA is needed for patients with 5β-reductase deficiency, as also is true for oxysterol 7α deficiency. We believe that primary bile acid treatment is necessary for 5β-reductase deficiency in infancy. After 6 months to 1year, follow-up treatment with UDCA or cessation of treatment might be considered. However, further study is needed, since phenotypes differ depending on specific genotypes of SRD5B1. To our knowledge, this is the first report of long-term outcome and efficacy of CDCA treatment in a group of patients with BASD from East Asia.
{kind=link}