Materials
Starting materials, chemicals and reagents used in this study (including acetophenone derivatives, thiourea, ammonia, iodine, 2-hydroxybenzaldehyde, nickel chloride, cobalt chloride, acetone, diethyl ether, chloroform, benzene) were supplied by Merck. Commercial ethyl alcohol was supplied by Ak Kimya, Aydın Adnan Menderes University, Turkey. Characterization of the synthesized compounds was done using an NMR spectrometer (Bruker GmbH DpX-400MHz Hing Performance Digital FT-NMR), FTIR spectrometer (Perkin Elmer 1600) and a melting point (m.p.) analyzer. The NMR chemical shifts (δ values) and coupling constants (J values) are reported in ppm and Hz, respectively.
Synthesis of 2-amino-4-thiazole derivatives
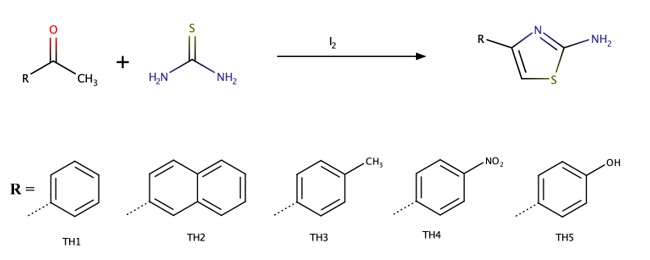
The aminothiazoles were synthesized by reacting the respective methyl-aryl ketone with thiourea in the presence of molecular iodine. A model reaction is given in Scheme 1.
Synthesis of 2-Amino-4-phenylthiazole ( TH1 )
Measured 12 g (0.1 mol) of acetophenone and 7.6 g (0.1 mol) of thiourea were transferred into a 100 mL flask. After, 14.5 g (0.05 mol) of iodine was added to the flask and mixed on a magnetic stirrer. The content of the flask was corked and placed in a water bath at 60\(℃\). After 24 hours, 50 ml of pure water was added and its cap was closed again and kept in the bath for a further one hour. At the end of 1 hour, the mixture in the flask was filtered while still hot. The yellow precipitate that remained on the filter paper was elemental sulfur while filtrate had a pH of 2. The filtrate was cooled and then 5% ammonia solution was added until a pH of 8 was attained. The resultant yellow precipitate formed was filtered, dissolved in ethyl alcohol and then crystallized by cooling in an ice bath. The crystals were washed with cold water and then dried in a vacuum desiccator.
Light-yellow; Yield: 14.6 g (83%); m.p: 149°C, mm = 176 g/mol (Fig. 1). Slightly soluble in petroleum ether, diethyl ether, pentane, n-hexane and CCl4; completely dissolved in acetone, benzene, CHCl3, DMF, DMSO, 1,4-dioxane, ethanol and methanol. 1H-NMR δ (ppm): 7.715 (d, 2H), 7. 362 (t, 1H), 7.253 (t, 2H), 7.035 (s, 2H), 5.997 (s, 1H); 13C-NMR δ (ppm): 168.645 (C-2), 150.308 (C-4), 135.382 (C-7), 128.909 (C-9 and C-11), 127.627 (C-10), 125.989 (C-8 and C-12), and 101.944 (C-5); FTIR (cm− 1): 3445 (N-H), 3310 (R-H thiazole ring), 3017 (R-H phenyl ring), 1610 (C = N), 1564 (C = C), 1505 (N-H), 655 (C-S-C).
Synthesis of 2-amino-4-(2-naphthyl)-thiazole ( TH2 )
Accurately weighed 17 g (0.1 mol) of 2-acetylnaphthalene and 7.6 g (0.1 mol) of thiourea were transferred into a 100 mL flask, followed by 14.5 g (0.05 mol) of iodine and 20 ml of ethyl alcohol. The mixture was stirred on a magnetic stirrer, corked and then placed in a water bath at 60 \(℃\). After 48 hours, the alcohol was evaporated. Exactly 50 ml of pure water was added, the cap was reclosed and kept in the bath for a further one hour. After, the mixture in the flask was filtered while still hot, cooled and then 5% ammonia solution was added to it until when a pH of 8 was attained. The light-yellow precipitate formed was filtered off, dissolved in alcohol, and then crystallized by cooling in an ice bath. The crystals obtained were filtered, washed with cold water and dried in a vacuum desiccator.
White; Yield: 17.63 g (78%); m.p: 226°C, mm = 118 g/mol (Fig. 2). Slightly soluble in petroleum ether, diethyl ether, pentane, n-hexane and CCl4, acetone, benzene, CHCl3, DMF, DMSO, but completely dissolved in 1,4-dioxane, ethanol and methanol. 1H-NMR δ (ppm): 8.119 to 7.426 (m, 7H), 7.133 (s, 1H), 7. 076 (s, 2H); 13C-NMR δ (ppm): 168.947 (C-2), 150.485 (C-4), 103.091 (C-5), 133. 899 (C-6), 133.030 (C-7), 132.984 (C-8), 128.752 (C-9), 128.569 (C-10), 128.340 (C-11), 128.203 (C-12), 127.005 (C-13), 126.472 (C-14) and 124.741 (C-15); FTIR (cm− 1): 3440 (N-H), 3307 (R-H thiazole ring), 3015 (R-H phenyl ring), 1613 (C = N), 1560 (C = C), 1501 (N-H), 657 (C-S-C).
Synthesis of 2-amino-4-(4-methylphenyl) thiazole ( TH3 )
Weighed 13.4 g (0.1 mol) of 4-methyl-acetophenone and 7.6 g (0.1 mol) of thiourea were added into a 100 mL flask. After adding 14.5 g (0.05 mol) of iodine, they were stirred using a magnetic stirrer, corked and then placed in a water bath at 60 oC for 24 hours. After, 50 ml of pure water was added to the mixture, corked and then kept in the bath for another one hour. The mixture was thereafter filtered while still hot, cooled and 5% ammonia solution was slowly added to it with continuous and rapid agitation until a pH of 8 was achieved and a dark yellow precipitate was observed on the sides of the beaker. The dark yellow precipitate formed (after 10 hours) was filtered out, dissolved in alcohol and crystallized by cooling in an ice bath.
Bright-yellow; Yield: 12.9 g (68%); m.p: 190°C, mm = 185 g/mol (Fig. 3). Slightly soluble in petroleum ether, diethyl ether, pentane, n-hexane and CCl4, acetone, benzene, CHCl3, DMF, DMSO but completely dissolved in 1,4-dioxane, ethanol and methanol. 1H-NMR δ (ppm): 7.680 (d, 2H), 7.165 (d, 2H), 6.996 (s, 2H), 6.911 (s, 1H), 2.250 (s, 3H); 13C-NMR δ (ppm): 168.544 (C-2), 150.382 (C-4), 136.821 (C-10), 132.750 (C-7), 129.967 (C-9 and C-11), 125.941 (C-8 and C-12), 101.019 (C-5), and 21.236 (C-13); FTIR (cm− 1): 3485 (N-H), 3325 (R-H in thiazole ring), 3098 (R-H in phenyl ring), 2962 (C-H, aliphatic), 1622 (C = N, in ring), 1555 (C = C), 1515 (N-H), 660 (C-S-C).
Synthesis of 2-amino-4-(4-nitrophenyl) thiazole ( TH4 )
To 16.9 g (0.1 mol) of 4-nitro-acetophenone and 7.6 g (0.1 mol) of thiourea in a 100 ml flask was added 14.5 g (0.05 mol) of iodine and 20 ml of ethyl alcohol. They were then stirred using a magnetic stirrer, corked and then the flask placed in a water bath at 60 \(℃\) for 72 hours. After, the alcohol was evaporated and 50 mL of pure water was added. The cap was closed again and kept in the bath at 95 \(℃\) for 1 hour. After, the mixture was filtered while still hot, cooled and 5% ammonia was added to it until the pH reaches 8 and an orange precipitate collected at the bottom of the beaker. The dark orange precipitate formed after 10 hours was filtered, dissolved in acetone and crystallized by addition of ether and cooling in an ice bath.
Dark-yellow; Yield: 10.83 g (49%); m.p: 221°C, mm = 225 g/mol (Fig. 4). Slightly soluble in petroleum ether, diethyl ether, pentane, n-hexane and CCl4, acetone, benzene, CHCl3, DMF, DMSO, and completely dissolved in in 1,4-dioxane, ethanol and methanol. 1H-NMR δ (ppm): 8.339 (d, 2H), 8.182 (d, 2H), 7.652 (s, 2H), 7.412 (s, 1H); 13C-NMR δ (ppm): 169.053 (C-2), 150.382 (C-4), 141.756 (C-7), 130.015 (C-9 and C-11), 197.587 (C-10), 124.420 (C-8 and C-12), and 107.036 (C-5); FTIR (cm− 1): 3493 (N-H), 3353, weak (R-H thiazole ring), 3101 (R-H phenyl ring), 1625 (C = N), 1582 (N = O), 1552 (C = C), 1515 (N-H), 675 (C-S-C).
Synthesis of 2-amino-4-(4-hydroxyphenyl) thiazole ( TH5 )
Aminothiazole derivative (TH5) was prepared by addition of 13.6 g (0.1 mol) of 4-nitro-acetophenone to 7.6 g (0.1 mol) of thiourea in a 100 mL flask. This was followed by 14.5 g (0.05 mol) of iodine and 20 ml of ethyl alcohol, which were then stirred on a magnetic stirrer, corked and transferred into a water bath at 60 \(℃\) for 48 hours. The alcohol was evaporated off and 50 ml of pure water was added to the mixture, capped and kept in the water bath at 95 \(℃\) for 1 hour. At the end of 1 hour, the mixture was filtered while still hot, cooled and 5% ammonia solution was added to the filtrate until a pH of 8 was reached and a white precipitate collects at the bottom of the beaker. The white precipitate formed (after 18 hours) was filtered and dissolved in alcohol and crystallized by cooling in an ice bath.
Greenish matt-colored; Yield: 12.1 g (63%); m.p: 192°C, mm = 213 g/mol (Fig. 5). Slightly soluble in petroleum ether, diethyl ether, pentane, n-hexane and CCl4, acetone, benzene, CHCl3, DMF, DMSO, but dissolved completely in 1,4-dioxane, ethanol and methanol. 1H-NMR δ (ppm): 9.473 (s, 1H), 7.628 (d, 2H), 6.973 (d, 2H), 6.762 (s, 2H), 6.695 (s, 1H); 13C-NMR δ (ppm): 168.507 (C-2), 150.586 (C-4), 130.571 (C-7), 115.678 (C-9 and C-11), 157.217 (C-10), 125.724 (C-8 and C-12), and 98.972 (C-5); FTIR (cm− 1): 3510 (N-H), 3410 (R-H in thiazole ring), 2800–3200, broadband (phenolic -OH), 1605 (C = N), 1592 (C = C), 1503 (N-H), 692 (C-S-C).
Synthesis of ligands (L1H and L2H) and complexes
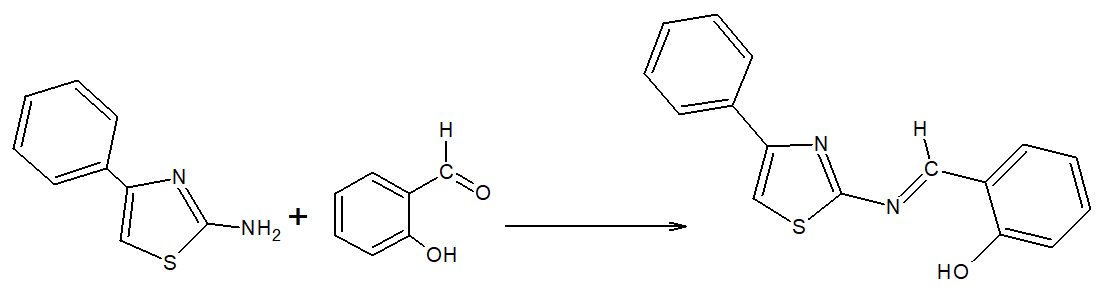
Synthesis of 4-phenyl-2-(2-hydroxymethylbenzilidenamino) thiazole ligand ( L1H )
L 1 H ligand was synthesized by reacting TH1 with 2-hydroxybenzaldehyde in the presence of acetic acid as a catalyst as shown in Scheme 2. Briefly, measured 8.8 g (0.05 mol) of TH1 was dissolved in 50 ml of alcohol, followed by drops of 6.2 g (0.05 mol) of 2-hydroxybenzadehyde solution in 20 ml of alcohol. After adding a few drops of acetic acid, the mixture was refluxed and stirred for 8 hours. The yellow-colored mixture turned red, was mixed using a magnetic stirrer, two-third of the solvent was evaporated and the remaining part was cooled by mixing again. Measured 50 ml of diethyl ether was to the mixture, covered and stirring continued overnight. At the end of this process, a yellow precipitate was formed at the bottom. The precipitate was filtered, and the filtrate was washed with cold alcohol and dried in the open air. L1H is a Schiff base that is suitable for metal complexation.
Yellow; Yield: 7.5 g (56%); m.p: 268°C, mm = 235 g/mol (Fig. 6). Insoluble in petroleum ether, diethyl ether, pentane and n-hexane, it is dissolved in acetone, DMF, DMSO and CHCl3. 1H-NMR δ (ppm): 11.583 (s, 1H), 9.267 (s, 1H), 7.831–6.865 (m, 9H, phenyl protons: 10–19), 6.345 (s, 1H); 13C-NMR δ (ppm): 168.345 (C-7), 164.753 (C-2), 161.031 (C-9), 155.068 (C-4), 149.761 (C-8), 117.557 (C-5), 136.538 (C-10), 135.668 (C-11), 134.692 (C-12), 132.328 (C-13), 129.026 (C-14), 128.866 (C-17), 128.675 (C-16 and C-18) and 120.135 (C-15 and C-19); FTIR (cm− 1): 3332 (R-H in thiazole ring), 3060 (R-H in phenyl ring), 1610, 1599 (C = N), 1483 (C = C), 699 (C-S-C).
Synthesis of 4-(4-methylphenyl)-2-(2-hydroxymethylbenzylideneamino) thiazole ligand ( L 2 H )
Another ligand L2H was synthesized in the same way as L1H but using TH3 with 2-hydroxybenzadehyde using acetic acid as the catalyst (Scheme 3). Measured 9.5 g (0.05 mol) of TH3 was dissolved in 50 mL of alcohol, followed by dropwise addition of 6.2 g (0.05 mol) of 2-hydroxybenzadehyde solution in 20 mL of alcohol. After adding a few drops of acetic acid, the mixture was taken to reflux, heated and stirred for 12 hours until the yellow-colored mixture turned red. Then the mixture taken from the reflux was mixed with a magnetic stirrer until 2/3 of the solvent evaporated and the remaining part was cooled by mixing again. To this, 50 ml of diethyl ether was added, covered and stirring was continued for a further 24 hours. At the end of the process, an orange precipitate was formed at the bottom which was filtered, and the filtrate washed with cold alcohol. It was dried in air.
Orange; Yield: 8.6 g (61%); m.p: 282°C, mm = 205 g/mol (Fig. 7). Insoluble in petroleum ether, diethyl ether, pentane and n-hexane, it is dissolved in acetone, DMF, DMSO and CHCl3. 1H-NMR δ (ppm): 11.577 (s, 1H), 9.222 (s, 1H), 7.839–6.746 (m, 8H, phenyl protons: 10–19), 6.316 (s, 1H), 2.256 (s, 3H); 13C-NMR δ (ppm): 168.116 (C-7), 164.638 (C-2), 160.993 (C-9), 155.061 (C-4), 149.753 (C-8), 117.534 (C-5), 138.223 (C-10), 136.088 (C-11), 132.321 (C-12), 131.924 (C-13), 129.026 (C-14), 129.278 (C-16 and C-18), 120.440 (C-15 and C-19), and 21.495 (C-21); FTIR (cm− 1): 3435 (R-H in thiazole ring), 3111 (R-H in phenyl ring), 2364 (C-H, aliphatic), 1662, 1537 (C = N), 1362 (C = C), 746 (C-S-C).
Synthesis of Nickel and Cobalt complexes of L1H and L2H ligands
Nickle and cobalt complexes were prepared from the synthesized ligands: L1H and L2H. To prepare the complexes (Scheme 4), the ligands were reacted with the respective divalent metal salts as follows.
For Ni(L1H)2, weighed 0.804 g (0.003 mol) of L1H was dissolved in 20 ml of acetone with heating. A green solution of 0.357 g (0.0015 mol) NiCl2.6H2O salt in 10 ml of absolute ethanol was dropwisely added to the mixture A light brown color was formed by the addition of a few drops of dilute NaOH solution. This mixture was refluxed for 8 hours to form an orange-colored precipitate. The hot solution was filtered, and the precipitate was washed sequentially with acetone, hot alcohol and water. The precipitate was dried in an oven at 100 oC.
Orange; insoluble in petroleum ether and CCl4, partially dissolved in acetone and methanol, completely dissolved in DMSO and 1,4-dioxane. FTIR (cm− 1): 3332 (R-H in thiazole ring), 3060 (R-H in phenyl ring), 1605, 1505 (C = N), 1438 (C = C), 698 (C-S-C).
For Co(L1H)2, measured 0.804 g (0.003 mol) of L1H was dissolved in 20 ml of acetone by heating the mixture. A blue solution of 0.359 g (0.0015 mol) CoCl2. 6H2O salt in 10 ml of absolute ethanol was added dropwisely, to give a light brown colored solution after the addition of a few drops of dilute NaOH solution. This mixture was refluxed for 8 hours to form a light green precipitate. The hot solution was filtered and the precipitate was washed sequentially with acetone, hot alcohol and water. The precipitate was dried in the oven at 100 oC.
Light-green; Insoluble in petroleum ether and CCl4, it partially dissolved in acetone and methanol, and completely in DMSO and 1,4-dioxane. FTIR (cm− 1): 3345 (R-H in thiazole ring), 3060 (R-H in phenyl ring), 1624, 1512 (C = N), 1455 (C = C), 699 (C-S-C).
For Ni(L2H)2, weighed 0.846 g of L2H was dissolved in 20 ml of acetone by heating. A green solution of 0.357 g (0.0015 mol) NiCl2.6H2O salt in 10 ml of absolute ethanol was added dropwisely, followed by a few drops of dilute NaOH solution to form a light brown solution. When this mixture was refluxed for 8 hours, a light green precipitate was formed. The hot solution was filtered, and the precipitate was washed sequentially with acetone, hot alcohol and water. The precipitate was dried in an oven at 100 oC.
Light green; Insoluble in petroleum ether and CCl4, partially dissolved in acetone and methanol, and completely in DMSO and 1,4-dioxane. FTIR (cm− 1): 3469 (R-H in thiazole ring), 2369 (C-H bending, aliphatic), 1605, 1501 (C = N), 1330 (C = C), 746 (C-S-C).
For Co(L2H)2, weighed 0.804 g of L2H was dissolved in 20 ml of acetone by heating. A blue solution of 0.359 g CoCl2.6H2O in 10 ml of ethanol and dilute NaOH solution were added dropwisely was to form a cloudy and pale-yellow solution. This mixture was refluxed for 8 hours to give a light green precipitate. The hot solution was filtered, and the precipitate washed sequentially with acetone, hot alcohol, and water. The precipitate was then dried in an oven at 100 oC.
Light green; Insoluble petroleum ether and CCl4, partially dissolved in acetone and methanol, and fully in DMSO and 1,4-dioxane. FTIR (cm− 1): 3217 (R-H in thiazole ring), 2231 (C-H bending, aliphatic), 1599, 1501 (C = N), 1312 (C = C).
Computational studies
The calculations were carried out using Density Functional Theory (DFT) method as implemented in the Gaussian 09 package (Frisch et al., 2016). The geometry optimizations were done using B3LYP functional with 631 + G(d,p) basis set (Becke, 1993; Miehlich et al., 1989). PCM model was used for the calculations in solvent (Bauernschmitt & Ahlrichs, 1996; Cossi & Barone, 2001).
Molecular docking studies
Molecular docking was used as a structural biology tool for assesment of the interaction between pairs of molecules (usually a receptor and a ligand). In this study, the two ligands synthesized were drawn into 2D models using Chemdraw software and thenconverted into 3D structures using Open Babel GUI version 2.3.2 (Open Bable GUI; Chris Morley, USA).
MGL Tools Autodock 4.0 package was used for docking interaction. The computational codes of the molecular targets were obtained from PubChem and Protein DataBank. Macromolecule sterilization, water, drug residues and foreign substances in it were cleaned with the software (Dilshad et al., 2022 ). Macromolecule + lıgand interactions were evaluated locally using grid-based atomic affinity potentials. Grid values used in Autodock software were taken as 80×80×80 A. At the end of the calculation carried out in the computer environment, the binding energies, hydrogen bonds and hydrogen bond lengths were calculated.
Using the molecular mechanics method to reduce the molecular charge of the ligand, the location of the atoms was roughly estimated and the ligand was placed in the force field. The new position obtained was used in the quasi-empirical optimization method, which balances the repulsion-pull potential of the ligand at the molecular mechanics’ position.
In the semi-empirical calculation, a new position file is created using PM6 as the pseudo potential. The resulting new position file creates the Density Functional Theory (DFT) position file. By using the DFT/B3LYP method and ((6-311G)(d.p)) basis sets, the coordinates with the lowest energy are written based on the bond lengths and bond angles of the structure. With the completion of the optimization, it was ensured that the ligand had the lowest energy, that is, the level that could make the best bond.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}