Artificial tubular molecular pockets bearing polar functionalities on their inner surface are useful model systems for understanding the mechanisms of protein-ligand interactions in living systems. We herein report a pillar[5]arene-derived molecular tube, [P4-(OH)BPO], whose endo conformational isomer endo-[P4-(OH)BPO] possesses an inwardly pointing hydrogen-bond (H-bond) donor (OH) in its deep cavity, a strong H-bond acceptor (C=O) on the predominantly hydrophobic inner surface, rendering it a perfect protein binding pocket mimetic. By measuring the binding affinity of this pocket-mimetic tube, we screened a library of various shape-complementary organic guests (1–38) resembling the fragment ligands in fragment-based drug design (FBDD). On the basis of the data for “fragment-pocket” complexes (1–38)⊂endo-[P4-(OH)BPO], two rationally designed “lead molecules” (39 and 40) were identified to be able to enhance binding affinity significantly by forming H-bonds with both the donor and acceptor of endo-[P4-(OH)BPO]. The described work opens new avenues for developing pillar[n]arene-derived protein binding pocket-mimetic systems for studies on protein-ligand interactions and mechanisms of enzymatic reactions.
Article
Pillar[5]arene-Derived endo-Functionalized Molecular Tube for Mimicking Protein-Ligand Interactions
https://doi.org/10.21203/rs.3.rs-145539/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Small molecules are the most common modality for new medicines, accounting for 67% of the FDA-approved drugs in 20191. Almost all current small-molecule drugs act by targeting disease-related proteins in the human body and regulating their activity2,3. As the interaction between a protein and a small molecule is often through binding of the molecule in a pocket on the protein surface, characterization of such binding pockets is important to elucidate the disease mechanisms and design new drug molecules4-6. Along with various methods, model studies using biomimetic receptors – artificially fabricated functional macrocyclic hosts – to mimic protein binding pockets are useful for understanding the nature of protein-ligand recognition7,8. During the past decades, the approach of fragment-based drug design (FBDD) has attracted increasing interest both in academia and pharmaceutical industry9-10. In FBDD, drug compounds were designed on the basis of protein-ligand bonding using fragment ligands with low complexity in chemical structures and low molecular weights9-10. It is well established that a protein binding pocket has an interior surface bearing both polar and non-polar functionalities for synergistic hydrogen bonding and hydrophobic interactions with a guest molecule4-5. In a reported survey of fragment-pocket complexes, hydrogen bonds (H-bonds) stabilize 89% of the fragment-pocket complexes, with 74% of the entries displaying one to three H-bonds10. Therefore, in order to imitate the microenvironment within protein binding pockets, the interior surface of the biomimetic receptors must possess polar functionalities for H-bond formation. To this end, there have been reports on pocket-shaped receptors, such as cavitands11, glucose receptor 212, aryl-extended calix[4]pyrrole receptors13, helical aromatic and oligoamide foldamers (Fig. 1a-e) 14,15. Jiang’s group recently made significant progress in transforming naphthalene-based tubular molecules into biomimetic tubes in which the inwardly directed functional groups improved their binding affinity and selectivity toward fragment ligands (Fig. 1f)16–18. As a class of tubular-shaped macrocyclic hosts, pillar[n]arenes19-21 could seat various functional substituents on their rims, for instance, amino acids or short peptides, for mimicking transmembrane channels22. Nonetheless, all of these rim-embedded polar functional substituents pointed outwards, and there has been no report on mounting polar functionalities pointing inwardly to the cavity cores of pillar[n]arenes. Herein, we report the creation of a pillar[5]arene-derived tubular molecular pocket with an inward-pointing hydroxyl group, endo-[P4-(OH)BPO] (Fig. 1g), which could unidirectionally interact with various small shape-complementary polar molecular guests through synergistic hydrophobic and hydrogen bonding interactions within its tubular cavity, mimicking a fragment-pocket in FBDD or protein-ligand interaction event in a protein binding pocket.
Formation of endo-[P4-(OH)BPO]. Our group has previously developed unique methods to grow functional substituents on the rims of pillar[n]arenes23. We conjectured that a pillar[n]arene-derived protein binding pocket mimetic could be realized through mounting a polar functionality on the rim of a pillar[n]arene, and then forcing it to point towards the tubular core either through steric effect or substrate-induced conformational change. We therefore started our work from adding a small polar functionality, hydroxyl group, to the rim of pillar[5]arene by creating a quaternary carbon atom through a nucleophilic addition reaction of pillar[4]arene[1]quinone (P4Q) with phenylmagnesium chloride (or phenyllithium) (Supplementary Information). The resultant product, pillar[4]arene[1]1-hydroxy-[1,1'-biphenyl]-4(1H)-one ([P4-(OH)BPO]), was characterized by 1H and 13C NMR spectroscopy, atmospheric pressure chemical ionization mass spectroscopy (APCI-MS), and single crystal X-ray diffraction analysis (Supplementary Information). The X-ray crystallography data of a single crystal obtained by slow evaporation of a solution of [P4-BPO(1-OH)] in acetone (Fig. 2a) clearly showed that ([P4-(OH)BPO]) has its phenyl group positioned outside the tubular frame, and its hydroxyl group pointing towards the core of the cavity, with an acetone molecule encapsulated inside the cavity through formation of a H-bond between the C=O group of acetone and the O–H group of [P4-(OH)BPO], which is a unambiguous evidence of the adoption of an endo conformational isomer, endo-[P4-(OH)BPO].
endo-[P4-(OH)BPO] as protein binding pocket mimetic. FBDD is an approach to develop potent small-molecule compounds starting from fragments binding weakly to target proteins9,10. Owing to its advantages such as saving experimental cost, offering diverse hits, and exhibiting multiple ways of binding, FBDD has been playing important roles in target-based drug discovery. In FBDD, fragment ligands are usually small molecules with low complexity in chemical structure, and H-bonds play an important role in stabilizing the fragment-pocket complexes9-10. For example, among the 462 unique fragment-pocket complexes investigated by Giordanetto, Shaw and co-workers, 92% of the fragments have at least one hydrogen bond formed with a protein, a structural water molecule, or a metal atom10. Given the role of H-bonds in fragment-pocket complexes, endo-[P4-(OH)BPO], with an inwardly pointing H-bond donor (-OH) at the bottom of its deep cavity, a strong H-bond acceptor (C=O) on the tubular inner wall of a predominantly hydrophobic surface, is naturally an appealing artificial protein binding pocket.
Binding of fragments to endo-[P4-(OH)BPO]. Recently, several biomimetic pockets with polar binding sites in their hydrophobic pockets have been disclosed11-18. Impressively, thermodynamics of interactions between biomimetic pockets and guests in water has been studied systematically by Jiang and co-workers16-18, which is highly valuable as the recognizations of ligands by protein binding pockets occur in aqueous environment in living systems. However, it is challenging to assess the contribution to pocket-ligand binding affinity “solely” from hydrogen bonding only through studies on the complexation in water. The reasons include: a) water molecules intend to bind the inner polar site of a synthetic receptor, and b) water molecules compete with a host in binding with a polar guest, weakening H-bond interaction between the host and guest16-18. In order to better understand both big picture and details of molecular recognition, investigation of pocket-fragment complexation in non-polar environment should be indispensably complimentary to the studies in aqueous solutions. Therefore, we initiated a thermodynamic study on the fragment-pocket binding abilities and selectivities of endo-[P4-(OH)BPO] with respect to various guest compounds in a non-polar solvent, CDCl3. In selecting guests for the study, ideal guest compounds resembling “fragment ligands” in FBDD are those not only geometrically complementary to the tubular binding pocket, but, more importantly, bearing H-bond acceptor(s) or donor(s) or both so that such a host-guest interaction could mimic a protein-ligand interaction event staged synergistically by both hydrogen bonding and hydrophobic interaction9,10. We selected an array of small-molecule fragment compounds for a “fragment library” which includes alkyl amines, alcohols, aldehydes, acids, esters, small heterocycles, and simple small molecules possessing bilogically important functional groups (Fig. 3). 1H NMR experiments were performed on all of the host-guest pairs at 1∶1 ratio in CDCl3 (Supplementary Information). The shifts of the proton signals of the complexed guests relative to the free species were used to evaluate the binding events. The binding constants (Ka) and binding free energies were determined by the 1H NMR titration method (Table 1)24. Generally, the energy of a single H-bond is somewhat between a van der Waals interaction and a fully covalent or ionic bond, but it varies depending on the nature of the donor and acceptor atoms, their geometries, and surrounding environments25,26. Thus, the “fragment-pocket” interaction of [P4-(OH)BPO] with a guest was expected to be affected by the number of H-bonds, angles of the hydrogen bond(s), nature of the donor and acceptor atoms, and the hydrophobic interaction between the inner surface of endo-[P4-(OH)BPO] and the guest25,26. In order to explore the correlations of the binding strength (Ka) and properties of guest fragments, molecular descriptors including the logarithm of the octanol:water partition coefficient (log P), dipole moment (μ), molecular volume (V), surface area (S), and asphericity Ωa) were collected16,27.
| Table 1. Fragment library compounds screened in fragment-pocket complexation | ||||||||
|
compound |
Ka (M-1)a |
R2 |
- ΔGb |
V (Å3)c |
Ωad |
S (Å2)e |
log Pf |
μ(D)g |
|
1 |
138.0±20.7 |
0.9957 |
11.8-12.6 |
129.80 |
0.18040 |
141.48 |
0.86 |
1.159 |
|
2 |
92.7±11.0 |
0.9942 |
10.9-11.5 |
177.01 |
0.21016 |
183.71 |
2.06 |
1.143 |
|
3 |
42.0±6.3 |
0.9880 |
8.9-9.6 |
224.21 |
0.22452 |
225.94 |
2.90 |
1.139 |
|
4 |
67.4±13.1 |
0.9977 |
9.9-10.9 |
123.07 |
0.18229 |
136.00 |
0.84 |
2.057 |
|
5 |
24.0±2.1 |
0.9936 |
7.7-8.1 |
170.27 |
0.21121 |
178.24 |
2.03 |
2.038 |
|
6 |
28.0±2.6 |
0.9941 |
8.0-8.5 |
217.47 |
0.22514 |
220.47 |
3.07 |
2.020 |
|
7 |
40.8±13.0 |
0.9957 |
8.2-9.9 |
115.11 |
0.18099 |
129.54 |
0.88 |
3.490 |
|
8 |
24.1±3.5 |
0.9987 |
7.5-8.2 |
162.30 |
0.20842 |
171.83 |
1.78 |
3.593 |
|
9 |
10.6±1.6 |
0.9998 |
5.4-6.2 |
209.51 |
0.22311 |
214.08 |
2.95 |
3.640 |
|
10 |
11.2±1.6 |
0.9989 |
5.6-6.3 |
147.78 |
0.16638 |
159.66 |
1.30 |
5.115 |
|
11 |
8.8±1.8 |
0.9987 |
4.8-5.9 |
195.01 |
0.20433 |
201.95 |
2.31 |
5.080 |
|
12 |
1.7±0.1 |
0.9996 |
1.2-1.5 |
242.19 |
0.22186 |
244.18 |
3.33 |
4.805 |
|
13 |
8.7±0.3 |
0.9992 |
5.3-5.4 |
123.99 |
0.13045 |
137.19 |
0.79 |
5.097 |
|
14 |
5.2±0.3 |
0.9982 |
3.9-4.2 |
171.15 |
0.19348 |
179.29 |
1.92 |
4.940 |
|
15 |
3.3±0.2 |
0.9989 |
2.8-3.1 |
218.36 |
0.21638 |
221.53 |
3.05 |
4.949 |
|
16 |
321.6±17.0 |
0.9999 |
14.2-14.4 |
83.08 |
0.11042 |
100.38 |
-2.06 |
4.853 |
|
17 |
22.5±2.2 |
0.9992 |
7.5-7.9 |
106.69 |
0.08171 |
120.67 |
-1.01 |
4.685 |
|
18 |
390.5±75.9 |
0.9957 |
14.3-15.2 |
106.79 |
0.08234 |
120.75 |
-1.55 |
4.961 |
|
19 |
241.5±22.9 |
0.9993 |
13.4-13.8 |
248.40 |
0.21017 |
247.71 |
1.50 |
5.259 |
|
20 |
109.4±16.1 |
0.9981 |
11.2-12.0 |
106.44 |
0.09492 |
121.96 |
-0.70 |
4.786 |
|
21 |
222.9±32.0 |
0.9985 |
13.0-13.7 |
130.15 |
0.13194 |
143.33 |
-0.19 |
4.391 |
|
22 |
103.9±3.6 |
0.9992 |
11.4-11.6 |
153.76 |
0.14146 |
163.59 |
0.32 |
4.446 |
|
23 |
58.6±3.2 |
0.9957 |
10.0-10.2 |
177.36 |
0.15542 |
184.78 |
0.83 |
4.412 |
|
24 |
123.5±13.6 |
0.9961 |
11.7-12.2 |
129.62 |
0.13367 |
142.39 |
-0.19 |
4.559 |
|
25 |
67.3±2.8 |
0.9955 |
10.3-10.5 |
247.79 |
0.17616 |
247.68 |
2.36 |
4.149 |
|
26 |
4.7±0.2 |
0.9993 |
3.7-3.9 |
138.11 |
0.05565 |
146.37 |
-0.64 |
4.631 |
|
27 |
ndh |
|
|
107.72 |
0.04061 |
119.30 |
0.46 |
2.470 |
|
28 |
ndh |
|
|
85.86 |
0.06253 |
99.95 |
0.75 |
3.745 |
|
29 |
83.0±15.1 |
0.9963 |
10.5-11.4 |
95.76 |
0.06253 |
106.64 |
-0.08 |
1.918 |
|
30 |
6.4±0.4 |
0.9987 |
4.4-4.8 |
90.78 |
0.06253 |
106.64 |
0.05 |
4.139 |
|
31 |
11.2±1.6 |
0.9989 |
5.6-6.3 |
101.33 |
0.07198 |
111.92 |
0.44 |
1.517 |
|
32 |
13.8±2.3 |
0.9989 |
6.1-6.9 |
124.95 |
0.10538 |
133.83 |
0.97 |
1.263 |
|
33 |
11.2±1.6 |
0.9989 |
5.6-6.3 |
136.16 |
0.12523 |
144.96 |
1.43 |
3.910 |
|
34 |
11.2±1.6 |
0.9989 |
5.6-6.3 |
185.70 |
0.16779 |
189.26 |
1.43 |
4.908 |
|
35 |
101.5±14.6 |
0.9983 |
11.1-11.8 |
102.60 |
0.03071 |
116.00 |
-1.35 |
5.552 |
|
36 |
52.4±9.0 |
0.9972 |
9.3-10.2 |
118.43 |
0.13632 |
132.34 |
0.87 |
4.572 |
|
37 |
28.6±3.2 |
0.9990 |
8.0-8.6 |
66.25 |
0.21003 |
84.87 |
-0.34 |
4.123 |
|
38 |
11.2±1.6 |
0.9989 |
5.6-6.3 |
159.13 |
0.18420 |
167.76 |
1.86 |
2.828 |
|
39 |
750.1±31.5 |
0.9999 |
16.3-16.5 |
163.20 |
0.15823 |
173.05 |
-1.31 |
2.743 |
|
40 |
1190±89.5 |
0.9999 |
17.4-17.7 |
139.89 |
0.14959 |
151.57 |
-2.52 |
6.032 |
aExperiments were performed in CDCl3 at 298K (Supplementary Information). bΔG = -RTlnKa. cCalculated based on optimized structures. dCalculated by the principal moments of inertia of guests. eCalculated by using Multiwfn program28. fValues were taken from references16,29,30. gCalculated with Gaussian 09 at the RB3LYP/6-31++G level of calcualtions31. hValues were too small to be determined.
Fragment-pocket complexation. We first assessed the binding between [P4-(OH)BPO] and fragments including alkyl amines, alcohols, aldehydes, acids, and esters (Table 1). The formation of fragment-pocket complexes between [P4-(OH)BPO] and the guests was evidenced by obvious upfield shifting of the proton signals of a-carbons of the guests (1-15) in the 1H NMR spectra of the fragment-pocket pairs in a 1∶1 molar ratio in non-polar solvent. As shown in the 1H NMR spectra (Fig. 4), upon mixing n-butyric acid (13) and [P4-BPO(1-OH)] in CDCl3, obvious upfield shifts for the methylene (Δδα = -0.26 ppm and Δδβ = -0.25 ppm) and the terminal methyl (Δδγ = -0.14 ppm) proton signals of 13 were observed. In addition, NOE correlations between protons (Hα) of 13 and the 1,4-dimethoxybenzene protons of [P4-BPO(1-OH)] were observed in the 2D NOESY spectrum (Supplementary Fig. S170). The 1H NMR and 2D NOESY data, together with Job plot (Supplementary Fig. S60) and APCI mass spectrum (peak m/z = 893.4014 for (13⊂[P4-BPO(1-OH)] + Li)+ (Supplementary Fig. S18), proved the formation of a 1:1 complex 13⊂[P4-BPO(1-OH)]. Similarly, alkyl amines (1–3) and alcohols (4–6) formed fragment-pocket complexes G⊂ [P4-BPO(1-OH)] (G = 1–6) with binding constants ranging from 40 to 140 M-1 and from 30 to 70 M-1, respectively. The binding constants between [P4-BPO(1-OH)] and aldehydes (7-9), esters (10-12) and acids (13-15) were smaller compared with those of amines (1-3) and alcohols (4-6). The large binding constants between the host and fragments 1-6 reflected stronger binding interaction originated from the H-bond accepting ability of the nitrogen atom of aliphatic amines or the oxygen atom of aliphatic alcohol32-34. As previously described by Smulders, Zarra and Nitschke27, higher μ and lower log P values meant more polarity of guests. Thus, it was not surprising to see that higher binding affinities correlate to higher μ and lower log P values in the cases of complexation in non-polar solvent CDCl3. Polar compounds tend to escape from non-polar solvent, and clutch H-bond donor in the deep pockets, which could explain the correlations between experimental values (Ka and ΔG) and calculated values (μ and log P) demonstrated by the guests 1-25 in the Table 1.
With the formation of fragment-pocket complexes confirmed, we wondered whether the host was in its endo conformation where the inward-pointing OH group forms a H-bond with a guest. Unambiguous evidence of endo conformation of [P4-BPO(1-OH)] in the complex was obtained from the X-ray diffraction analysis. The single crystal structure of the complex 13⊂endo-[P4-BPO(1-OH)] (Fig. 2c) revealed that 13 is encapsulated in the host pocket with its alkyl chain in the hydrophobic tubular region of the host cavity, and, unmistakably, with its carbonyl oxygen H-bonded to the inwardly pointing OH group in the deep cavity of the host, with the CO···HO distance of 1.941 Å, perfectly in the range of a typical H-bond length35. Thus, the complex 13⊂endo-[P4-BPO(1-OH)] is synergistically stabilized by both hydrogen bonding and hydrophobic interactions. We also obtained crystal structures of a few representative fragment-pocket complexes G⊂endo-[P4-BPO(1-OH)] (G = 9, 17, 21, 24, 25, 31, and 35) in which the carbonyl oxygen is bonded to the OH group of endo-[P4-BPO(1-OH)] in the deep pocket (Fig. 2b, 2d-i). Consequently, we could deduce that in the fragment-pocket complexes 1-25⊂[P4-BPO(1-OH)], [P4-BPO(1-OH)] was in an endo conformation with its OH group inwardly pointing to interact with H-bond acceptor of a guest fragment.
Amides are commonly seen in drug molecules36. Amide functional groups have weak basic and acidic possibilities37. The role of a H-bond between an amide guest and the hydroxyl of [P4-(OH)BPO] in stabilizing a fragment-pocket complex is significant37, which was reflected in our model fragment-pocket binding study: Complexes G⊂endo-[P4-(OH)BPO] (G = 16–25, Table 1) were found to have much larger Ka values than G⊂endo-[P4-(OH)BPO] (G = 1–15, 26–38, Table 1). Besides H-bonding, hydrophobic interaction also plays a significant role in stabilizing the fragment-pocket complexes in which the hydrophobic fragments of the ligands are energetically favored at the interface of the host pocket38,39 Thus, the binding mode of an amide fragment in a “fragment-pocket” complex is determined not only by the strength of amide carbonyl-hydroxyl (CO···HO) H-bond, but also by the hydrophobic interaction between the inner cavity of the host and the guest chain38,39. This was well reflected in complexes 21⊂endo-[P4-(OH)BPO] and 24⊂endo-[P4-(OH)BPO] where alkyl chains of guests 21 and 24 were trapped in the host pocket albeit they were connected to different points of amide functional group (either to the amino N or the carbonyl C atoms, respectively), as shown by the single crystal X-ray structures (Fig. 2e and 2f). The hydrophobic interaction between the predominantly hydrophobic inner surface of the pocket and the alkyl chain of the fragment determined the binding modes of these two complexes. As 21 was more polar (higher μ and lower log P value) than 24 (Table 1), the Ka value of 21⊂endo-[P4-(OH)BPO] doubled that of 24⊂endo-[P4-(OH)BPO] (Table 1). As amide 25 has two butyl groups connected to the amino N and the carbonyl C, respectively, two possible binding modes for 25 exist with either of the two butyl groups confined in the cavity of endo-[P4-(OH)BPO]. The 1H NMR data (Supplementary Fig. S21, ) and the crystal structure of 25⊂endo-[P4-(OH)BPO] (Fig. 2g) indicated that the amine subgroup of 25 was left outside of the binding pocket, possibly owing to the difference in charge density and polarity between the N and C atoms associated with the C=O (Mulliken charges calculated to be -0.518556e and -0.417205e for the N and C atoms, respectively) (Supplementary Information). As a consequence, the solvation interaction between the solvent-exposed N-butyl chain and the “nonpolar” solvent (CDCl3) brought down the stability of 25⊂endo-[P4-(OH)BPO]40, as evidenced by its lower Ka than those of the 21⊂endo-[P4-(OH)BPO] and 24⊂endo-[P4-(OH)BPO].
Heterocyclic rings are predominant architectural components of pharmaceuticals and allow for variable biological interactions otherwise hard to achieve41,42. The binding of five membered heterocycles (26-34), non-aromatic and aromatic, to endo-[P4-BPO(1-OH)] were thus studied. The obvious upfield shifts of the proton signals of the five membered heterocyclic guests (26-34) in the 1H NMR spectra in CDCl3 upon mixing with [P4-BPO(1-OH)] evidenced the formation of the corresponding host-guest complexes (Table 1). Unambiguous evidence of an examplar heterocyclic fragment-pocket complex was provided by the single crystal structure of 30⊂endo-[P4-BPO(1-OH)] which revealed that thiazole 30 is cramped in the tubular cavity of endo-[P4-BPO(1-OH)] with its N atom forming a H-bond with the -OH group of the host (Fig. 2h). Among “screened” heterocyclic fragments, imidazole 29 formed fragment-pocket complex with a higher bonding constant (Table 1), which implied that 29 might use both of its H-bond acceptor (=N) and donor (-NH) in the interaction with the pocket.
Sulfoxide, oxirane, nitrile and nitro group frequently appear in pharmaceuticals43-47. We therefore included fragments dimethyl sulfoxide (DMSO) (35), 1-nitropropane (36), acetonitrile (37) and 2-butyloxirane (38) in our exploration of fragment-pocket interaction study. DMSO molecule is dipolar aprotic with a strong polar sulfoxide group and two hydrophobic methyl moieties43,44, which facilitate the formation of a stable complex 35⊂endo-[P4-BPO(1-OH)] (Fig. 2f). With a nitro group that forms H-bond with a -OH group45, plus a propyl chain to reach the hydrophobic area in the host’s cavity, fragment 36 formed stable 36⊂endo-[P4-BPO(1-OH)] as expected. Nitrile group plays efficacious roles of in drug molecules46, so acetonitrile (37), with simple structure (linear N≡C−C skeleton) and small size (C−N distance 1.16 Å), was selected as a nitrile fragment. The H-bond formed between C≡N nitrogen and -OH of the host was not strong, and the hydrophobic methyl did not contribute much binding affinity, which result in a low binding constant for 37⊂endo-[P4-BPO(1-OH)]. 2-Butyloxirane (38) formed 38⊂endo-[P4-BPO(1-OH)] with a low Ka possibly owing to the weak H-bond between OH of the host and the oxirane oxygen47.
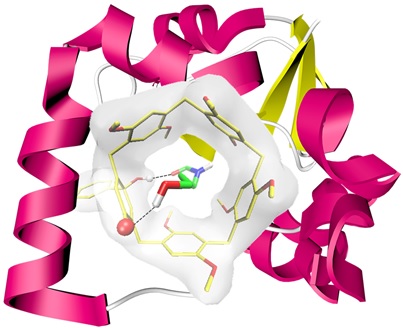
Fragment-based drug design (FBDD). Encouraged by the aforementioned results of the fragment-pocket complexes G⊂endo-[P4-BPO(1-OH)] (G = 1–38), we attempted a model FBDD aiming at “lead molecules” to bind the “target” pocket – endo-[P4-BPO(1-OH)]. Among the fragments 1-38, amides 16, 18, 19 and 21, with amine alkyl chains, gave binding constants greater than 200 M-1, so these N-alkyl substituted formamides and acetamides were designated as the screening-generated “hit molecules”. Given that all of the fragments 1-38 are monofunctional, and do not have structural handle to utilize the H-bond acceptors on the inner surface of endo-[P4-BPO(1-OH)] cavity, bifunctional amides 39 and 40 possessing an H-bond donor at the other end of the molecules were designed for the “target” endo-[P4-BPO(1-OH)]. We were delighted to see that the binding constants of designed bifunctional “lead molecules” 39 and 40 were enhanced by more than 13 and 20 folds, respectively, compared to their monofunctional analog 23 (Table 1). We were delighted to see that 40 achieved the highest binding strength among all of the fragments listed in Table 1 with a Ka of 1.2 × 103 [M-1]. The stronger binding of 40 with endo-[P4-BPO(1-OH)] than that of 39 could be partially explained by the higher diploe moment and lower log P of the former than the latter (Table 1). In the 1H NMR spectra of both complexes (Supplementary Fig. S45 and S46), significant upfield shifting of methylene proton signals of the bound guest was observed, implying that the guests were embraced inside the pocket cavity. Despite extensive efforts, crystalization of the fragment-pocket complexes 39 or 40⊂endo-[P4-BPO(1-OH)] was not successful. Nonetheless, computational docking of 39 and 40 to the pocket of endo-[P4-BPO(1-OH)] (Fig. 5) clearly demonstrated that the “lead molecules” were bound to the pocket cavity through a H-bond between amide C=O of the fragment and OH in the deep pocket with O···H distance of 1.573 Å and 1.597 Å, respectively, and, remarkably, the second H-bond between the fragment’s OH and carbonyl O on the inner wall of pocket cavity with bond length, 1.955 Å and 1.865 Å respectively (Fig. 5).
Complexation through “induced-fit”. It is generally believed that the binding of a ligand to a conformationally free protein is mechanistically via “conformational selection”, whereby a ligand selectively binds to a form of the protein pocket, or via “induced fit”, whereby a ligand binds to a predominantly free conformation of a protein, followed by a conformational change of the protein to form a preferred protein-ligand complex49,50. Theoretically, the rim-bound hydroxyl group in conformationally free [P4-(OH)BPO] should be able to park on either inside or outside the tubular wall to result in endo-[P4-BPO(OH)] or exo-[P4-BPO(OH)]23. In solution, [P4-(OH)BPO] exists in a rapid equilibrium between endo-[P4-(OH)BPO] and exo-[P4-(OH)BPO] isomers, as evidenced by the single set of proton signals in its 1H NMR spectra in various deuterated solvents, such as dichloromethane-d2, chloroform-d3, acetone-d6, acetonitrile-d3, DMF-d4 and DMSO-d6 (Supplementary Fig. S4), most probably owing to interconversion between the endo- and exo-conformational isomers in a rate faster than the NMR timescale. We therefore deduced that formation the fragment-pocket complexes G⊂endo-[P4-(OH)BPO] (G = 1–40) could be an “induced fit” process where a fragment guest passing through the host tube clutches the freely moving OH through H-bond interaction, and, as a consequence, freezes conformationally free [P4-(OH)BPO] into an endo-complex. Based on this assumption, we conjectured that an exo-[P4-(OH)BPO] conformational isomer could be also “frozen” out by non-polar fragment guest geometrically complementary. As expected, exo-[P4-(OH)BPO] conformational isomer was “frozen” out by solvent-induced crystallization in hexane, and was confirmed by 1H and 2D NOSEY NMR spectra in CDCl3 (Supplementary Fig. S47 and S175) and unambiguously single crystal X-ray structure (Fig. 6) in which the phenyl group linked to the quaternary carbon atom lands on the inner side of the tubular core, leaving the hydroxyl group pointing outside, a hexane molecule is imprisoned in the tubular cavity through solely hydrophobic interaction (Fig. 6).
In summary, we have fabricated a pillar[5]arene-derived molecular tube [P4-(OH)BPO]. With an inwardly pointing hydrogen bond donor (OH) in the deep cavity and a hydrogen bond acceptor (C=O) on the inner surface, plus a hydrophobic tubular inner wall, the endo-conformational isomer, endo-[P4-BPO(1-OH)], renders itself a perfect artificial protein binding pocket. On the basis of the study on the binding behavior of various FBDD “fragment ligands” inside the “pocket” of endo-[P4-(OH)BPO], “lead molecules” were rationally designed with their binding constants greatly enhanced compared to the screening-generated “hit moleclues”. The further work in this direction could be useful in FBDD and artificial protein-ligand binding models to facilitate our understanding of complex molecular interactions in living systems.
General methods. Unless otherwise noted, all commercial reagents and solvents were used without purification. Separation by flash column chromatography was performed on silica gel (200-300 mesh). 1H and 13C NMR spectra were recorded on Bruker AVANCE NEO 400 NMR spectrometer in CDCl3 at 298K. Atmospheric pressure chemical ionization (APCI) mass spectra were recorded on a Thermo Scientific Q Exactive Focus (UltiMate 3000 HPLC) mass spectrometer. Single crystal X-ray diffraction data were collected at 100 K on a Rigaku-Oxford Diffraction SuperNova dual source diffractometer (Cu at Zero equipped with an AtlasS2 CCD using Cu Kα radiation). The data collected were processed by CrysAlisPro. The structures were solved by direct methods using Olex2 software, and the non-hydrogen atoms were refined anisotropically with SHELXL-2018 using a full-matrix least-squares procedure based on F2. The weighted R factor, wR and goodness-of-fit S values were obtained based on F2.The hydrogen atom positions were fixed geometrically at the calculated distances and allowed to ride on their parent atoms. Crystallographic data for the structures disclosed in this paper have been deposited at the Cambridge Crystallographic Data Center (CCDC).
NMR experiments. 1H NMR experiments were performed on all of the host–guest pairs at a 1∶1 ratio, and the peak shifts of the complexed host and guest relative to the free species were used to access complexation. In the determination of binding constants and stoichiometries of complexes G⸦[P4-(OH)BPO] (G = 1–41), 1H NMR titration25 was performed in CDCl3 with the host concentration of 10.0 mM and varying concentrations of the guest at 298K. By a non-linear curve-fitting method, the association constant between guest and host was calculated from the equation:

where Δδ is the chemical shift change of Ha on host at [guest]0, Δδ∞ is the chemical shift change of Ha when host is completely complexed, [host]0 is the fixed initial concentration of host, and [guest]0 is the varying concentrations of guest.
Calculations. Quantum chemical calculations were performed using Gaussian 1632. Fragment library compounds and designed “drug molecules” were calculated by RMO62X 6-311G (d,p) level of theory, and vibrational frequency calculations have be carried out to verify that all optimized geometries are true minima. Molecular volume (V) and surface area (S) of guest moleculars were calculated using Marching Tetrahedron algorithm via Multiwfn program29. Dipol momnetvalues were calculated using Gaussian 09 at the RB3LYP/6-31++G level of calcualtion.
Asphericity(Ωa) was defined as

IA, IB and IC are the principal moments of inertia of a molecule28. Mulliken charges were calculated by Gaussian 09 at PM6-D3 level. Molecular docking of G⸦[P4-(OH)BPO] (G = 39 or 40) was performed with AutoDock Vina48.
Data availability
All data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding authors upon reasonable request. Crystallographic data for the structures reported in this article have been deposited in the Cambridge Crystallographic Data Centre, under CCDC numbers: 2052256, 2052257, 2052258, 2052259, 2052260, 2052261, 2052262, 2052264 and 2052265. Copies of the data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/structures. All crystallographic data are available in supporting information.
Acknowledgements
This study was financially supported by National Key Research and Development Program of China (2017YFA0206500), National Natural Science Foundation of China (21871281 and 21902176), Shanghai Sailing Program (19YF1452900), Youth Innovation Promotion Association, CAS (2020291) and Science and Technology Commission of Shanghai Municipality (18DZ1100403). We thank the NMR Core Facility, School of Life Science and Technology, ShanghaiTech University for the assistance on NMR analyses.
Author contributions
Z.W., Y.A.L., W.-B.H., H.Y. and K.W. conceived the project. Z.W., T.C., H.L. and X.-L.Z. conducted the experiments and data analyses, including synthesis and characterization of [P4-BPO(1-OH)], NMR experiments, crystal growths, quantum chemical calculations of the fragments, calculation of Mulliken charges, molecular docking, and single-crystal X-ray crystallographic analysis. Z.W., Y.A.L. and K.W. prepared the manuscript with contributions from all authors.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://........................................
Correspondence and requests for materials should be addressed to K.W.
Reprints and permissions information is available at www.nature.com/reprints.
- Jarvis, L. M. The new drugs of 2019. C&E News. 98, 30-36 (2020).
- Gao, M. & Skolnick, J. A Comprehensive survey of small-molecule binding pockets in proteins. PLOS Comput. Biol. 9, e1003302 (2013)
- Stank, A., Kokh, D., B., Fuller, J. C. & Wade, R. C. Protein binding pocket dynamics. Chem. Res. 49, 5, 809–815 (2016).
- Zheng, X., Gan, L., Wang, E. & Wang, J. Pocket-based drug design: Exploring pocket space. AAPS J. 15, 228–241 (2013).
- Coleman, R. G. & Sharp, K. A. Protein pockets: Inventory, shape, and comparison. J. Chem. Inf. Model. 50, 589–603 (2010).
- Weskamp, N., Hullermeier, E. & Klebe, G. Merging chemical and biological space: Structural mapping of enzyme binding pocket space. Proteins: Struct., Funct., Bioinf. 76, 317–330 (2009).
- McFedries, A., Schwaid, A. & Saghatelian, A. Methods for the elucidation of protein-small molecule interactions. Biol. 20, 667–673 (2013).
- Smith, B. D. (Ed.) Synthetic Receptors for Biomolecules: Design Principles and Applications (The Royal Society of Chemistry, Cambridge, 2015)
- Erlanson, D. A. & Jahnke W. () Fragment-based Drug Discovery. Lessons and Outlook (Wiley-VCH, Weinheim, Germany, 2016).
- Giordanetto, F., Jin, C.; Willmore, L., Feher, M. & Shaw, D. E. Fragment hits: What do they look like and how do they bind? Med. Chem. 62, 3381–3394 (2019).
- Sara M. Butterfield. S. M. & Rebek. J. A synthetic mimic of protein inner space: Buried polar interactions in a deep water-soluble host. Am. Chem. Soc. 128, 15366-15367 (2006).
- Tromans, R.A. et al. A biomimetic receptor for glucose. Nature Chem. 11, 52–56 (2019).
- Peñuelas-Haro, G. & Ballester, P. Efficient hydrogen bonding recognition in water using aryl-extended calix[4]pyrrole receptors. Chem. Sci. 10, 2413–2423 (2019).
- Kim, K. M. et al. Template-directed quantitative one-pot synthesis of homochiral helical receptors enabling enantioselective binding. Chem. Int. Ed. 59, 22475-22479 (2020).
- Chandramouli, N. et al. Iterative design of a helically folded aromatic oligoamide sequence for the selective encapsulation of fructose. Nature Chem 7, 334–341 (2015).
- Yao, H. et al. Molecular recognition of hydrophilic molecules in water by combining the hydrophobic effect with hydrogen bonding. Am. Chem. Soc. 140, 13466–13477 (2018).
- Huang, G., Valkonen, A., Rissanen, K. & Jiang, W. endo-Functionalized molecular tubes: Selective encapsulation of neutral molecules in non-polar media. Chem Commun. 52, 9078–81 (2016).
- Ma, Y.-L. et al. Biomimetic recognition of organic drug molecules in water by amide naphthotubes. CCS Chem. 2020, 2, 1078–1092 (2020).
- Ogoshi, T., Kanai, S., Fujinami, S., Yamagishi, T. & Nakamoto, Y. para-Bridged symmetrical pillar[5]arenes: Their Lewis acid catalyzed synthesis and host–guest property. Am. Chem. Soc. 130, 5022-5023 (2008).
- Xue, M., Yang, Y., Chi, X., Zhang, Z. & Huang, F. Pillararenes, A new class of macrocycles for supramolecular chemistry. Chem. Res. 45, 1294-1308 (2012).
- Song, N. & Yang, Y.-W. Applications of pillarenes, an emerging class of synthetic macrocycles. China Chem. 57, 1185-11989 (2014).
- Hu, X.-B., Chen, Z., Tang, G., Hou, J.-L. & Li, Z.-T. Single-molecular artificial transmembrane water channels. Am. Chem. Soc. 134, 8384−8387 (2012).
- Xie, C.-D. et al. Synthesis of pillar[n]arene[5-n]quinines via partial oxidation of pillar[5]arene. J. Chem. 33, 379-383 (2015).
- Pall, T. Determining association constants from titration experiments in supramolecular chemistry Soc. Rev. 40, 1305–1323 (2011).
- Wendler, K., Thar, J., Zahn, S. & Kirchner, B. Estimating the hydrogen bond energy. Phys. Chem. 114, 9529–9536 (2010).
- Jeffrey, G.-A. An introduction to hydrogen bonding (Oxford university, New York, 1997).
- Smulders, M. M. J., Zarra, S. & Nitschke, J. R. Quantitative understanding of guest binding enables the design of complex host–guest behavior. Am. Chem. Soc. 135, 7039–7046 (2013).
- Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. Comput. Chem. 33, 580–592 (2012).
- Rumble, J. R., Lide, D. R. & Bruno, T. J. CRC Handbook of Chemistry and Physics, 100th ed. (CRC Press, Boca Raton, 2019).
- https://scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf (accessed Dec 6, 2020).
- Frisch M. J. et al.Gaussian 16 Revision A 03 (Gaussian Inc., Wallingford, 2016)
- Spencer, J.-N., Wolbach, W.-S., Hovick, J.-W., Ansel, L. & Modarress, K. J. Hydrogen bonding by alcohols and amines, Solution Chem. 14, 805–814 (1985).
- Mazur, K., Bonn, M. & Hunger, J. Hydrogen bond dynamics in primary alcohols: A femtosecond infrared study. Phys. Chem. B 119, 1558–1566 (2015).
- Lommerse, J. P. M., Price, S. L. & Taylor, R. Hydrogen bonding of carbonyl, ether, and ester oxygen atoms with alkanol hydroxyl groups, Comput. Chem. 18, 757–774 (1997).
- The typical length of a hydrogen bond in water is 97 Å (197 pm). Bond distances (measured from the hydrogen atom) range from 1.5 to 2.6 Å, or from 2.4 to 3.5 Å separate the two nonhydrogen atoms in a hydrogen bond. See Berg, J. M., Tymoczko J. L. & Stryer, L. Biochemistry. 5th ed. (Freeman. W. H. Press, New York, 2002).
- Comer, J. E. A. Ionization constants and ionization profiles. In Comprehensive Medicinal Chemistry II Testa, B. & van de Waterbeemd, H. eds. (Elsevier Press, Amsterdam, 2007).
- Habermann, S. M. & Murphy, K. P. Energetics of hydrogen bonding in proteins: A model compound study. Protein Sci. 5, 1229–1239 (1996).
- Chema, D., Eren, D., Yayon, A., Goldblum, A. & Zaliani, A. Identifying the binding mode of a molecular scaffold. Comput. Aided Mol. Des. 18, 23–40 (2004).
- Motiejunas, D. & Wade, R. C. Structural, energetic, and dynamic aspects of ligand-receptor interactions. In Comprehensive Medicinal Chemistry II, Vol 4 Mason, J. S. (Ed.) ( Press, Amsterdam, 2007).
- Naïm, M. et al. Solvated interaction energy (SIE) for scoring protein−ligand binding affinities. 1. Exploring the parameter space. Chem. Inf. Model. 47, 122–133 (2007).
- Baumann, M., Baxendale, I. R., Ley, S. V. & Nikbin, N. An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. Beilstein J. Org. Chem. 7, 442–495 (2011).
- Dalvie, D. K. et al. Biotransformation reactions of five-membered aromatic heterocyclic rings. Res. Toxicol. 15, 269–299 (2002).
- Surur, A. S., Schulig, L. & Link, A. Interconnection of sulfides and sulfoxides in medicinal chemistry, Pharm. 352, 1800248 (2019).
- Pacak, P. Polarizability and molecular radius of dimethylsulfoxide and dimethylformamide from refractive index data. Solution Chem., 16, 71–77 (1987).
- Nepali, K., Lee, H. Y. & Liou, J. P. Nitro-group-containing drugs. Med. Chem. 62, 2851−2893 (2019).
- Fleming, F. F., Yao, L. H., Ravikumar, P. C., Funk, L. & Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. Med. Chem. 53, 7902–7917 (2010).
- Delost, M. D., Smith, D. T., Anderson, B. J. & Njardarson, J. T. From oxiranes to oligomers: Architectures of U.S. FDA approved pharmaceuticals containing oxygen heterocycles. Med. Chem. 61, 10996–11020 (2018).
- Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Comput. Chem. 31, 455–461 (2010).
- Hammes, G. G., Chang, Y. C. & Oas, T. G. Conformational selection or induced fit: A flux description of reaction mechanism. PNAS. 106, 13737–13741 (2009).
- Vogt, A. D. & Di Cera, E. Conformational selection or induced Fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894−5902 (2012).
There is NO Competing Interest.
- Supportinginformation.pdf
Pillar[5]arene-Derived endo-Functionalized Molecular Tube for Mimicking Protein-Ligand Interactions
- GraphicalAbstract.jpg
{kind=link}