Workflow Analysis and Data Description
The analytical workflow is shown in Figure 1. First, we evaluated the differences in immune cell infiltration and tumor microenvironments of sarcoma tissues and normal, adjacent tissues. Next, we characterized the significant module associated with CD8 T cells by WGCNA, and hub genes were identified and validated. Samples from 263 patients with sarcoma and two samples of normal, adjacent tissues were obtained from the TCGA database for further analysis.
The Percent of Immune Cells in Sarcoma Samples and Clinical Correlation
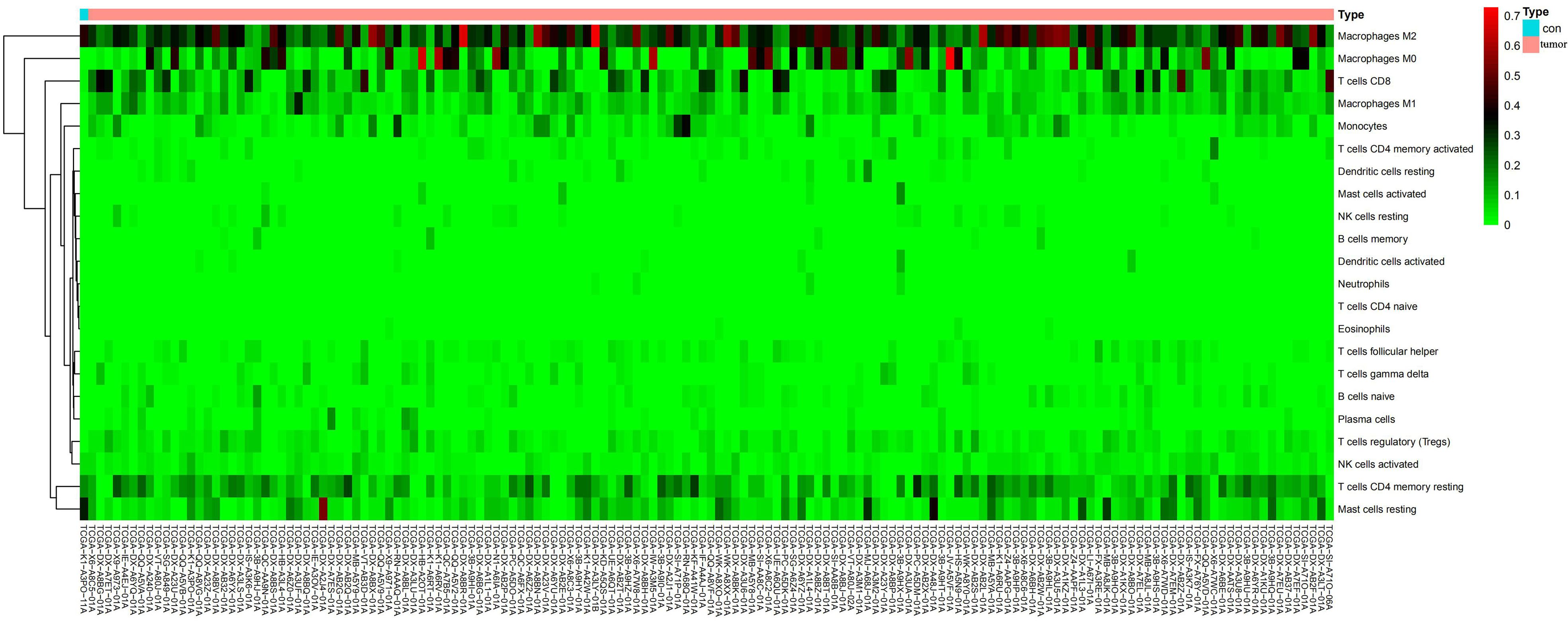
CIBERSORT is an algorithm, based on the machine-learning, highly sensitive and specific discrimination of 22 human immune cell phenotypes in several cancer types[36, 37]. In this study, we used CIBERSORT algorithm to assess the composition of immune cells in sarcoma and normal samples (Figure 2A-B, Supplementary Figure 1). Macrophages were the predominant immune cell type in sarcoma tissues. In addition, the fraction of CD8 T cells was negatively associated with M0 macrophages (R = -0.43) and M2 macrophages (R = -0.41), and positively associated with M1 macrophages (R =0.51) and follicular, helper T cells (R = 0.6)(Figure 2C).We performed a Kaplan-Meier survival analysis to evaluate the correlation between the 22 immune cell subtypes and the prognosis of patients from whom the tumors were acquired. The fraction of activated dendritic cells (p = 9.293e-04), resting dendritic cells (p = 0.031), M2 macrophages (p = 1.668e-04), neutrophils (p = 0.046), and plasma cells (p = 0.031) were significantly different among disease types. The fractions of activated dendritic cells (p = 0.04) and M2 macrophages (p = 0.023) were significantly different among disease recurrence, and the fraction of activated dendritic cells (p = 0.021), resting NK cells (p = 0.007), and follicular helper T cells (p = 0.028) were also significantly different among total necrosis percent (Supplementary Figure 2). In addition, the fractions of activated NK cells (p = 0.049), CD8 T cells (p < 0.001), regulatory T cells (p = 0.003), M0 macrophages (p = 0.002), and M1 macrophages (p = 0.038) were significantly correlated with overall survival (Figure 3).
Immune-, Stroma- and Estimate- Scores Correlated with Clinical Parameters
To evaluate the microenvironment infiltration of immune and stromal, we applied the The “ESTIMATE” package in R to match and calculate the Immune-, Stromal-, and Estimate- scores of 263 patients. As shown in Table 1, the stromal scores ranged from -1336.63 to 2476.22, the Immune scores ranged from -1722.08 to 3499.2, and the Estimate scores ranged from -2977.43 to 5336.41. The “survival” package in R was utilized to analyze the correlations of Estimate, Immune, and Stromal scores with overall survival (Figure 4). The patients with tumors that had high Estimate, Immune, and Stromal scores had a significantly better prognosis than patients with tumors that had low Estimate, Immune, and Stromal score group (p = 0.004, p = 0.007, p = 0.017, respectively). Furthermore, the relationship among Estimate-, Immune-, Stromal- scores and clinical parameters were evaluated (Supplementary Figure 3). The clinical parameters (disease type, gender) significantly increased as Estimate-, Immune-, and Stromal- scores increased (p < 0.05).
Identification of DEGs and Clinical Correlation
To further investigate the function of pivotal gene in microenvironment infiltration, the gene expression profiles were differentiated into differentiate the two groups (high vs. low, Supplementary Figure 4). Using the Immune scores, a total of 2199 DEGs between the high-score and low-score group were identified in which 1070 genes were upregulated, and 1129 genes were downregulated. Similarly, for stromal scores, 995 DEGs were upregulated and 1498 DEGs were downregulated between the high-score and low-score groups. The genes differentially upregulated and downregulated in the high vs. low immune and stromal scores groups were further analyzed. Of these 1509 genes, 729 were upregulated and 780 were downregulated (Supplementary Figure 5).
Weighted Gene Co-expression Network Construction and Module Preservation Analysis
WGCNA focused on transforming gene expression data into co-expressed module and screening out hub genes, providing insights into correlation between genes in different samples[26]. After validation, the 1509 DEGs were used to create a co-expression network using WGCNA. The analysis was performed as previously described [27]. Briefly, the gene expression profile matrix of all samples was transformed into a Pearson’s correlation coefficient matrix, and the distribution of connections among these genes indicated that the profile met the criteria of a scale-free network. Then we constructed network based on the scale-free network (Figure 5A). We assigned a power value of 4 with a 0.8 degree of independence (Supplementary Figure 6, Figure 5B). The size of the seven modules ranged from 21 to 338 genes. Genes that were not co-expressed were assigned to the grey module, and were not further analyzed. Furthermore, the results of module stability analysis demonstrated that the Zsummary.qual for module preservation of gold modules was < 5, and the Zsummary statistic for module preservation of the blue, green, yellow, turquoise, and brown modules was > 10 (Figure 5C, Supplementary Table S1). Based on these data, the gold module was not considered stable in these analyses, and it was not used in subsequent analyses.
Identifying Significant Modules and Module Functional Annotation
The association of the six modules with types of immune cell infiltration was analyzed (Figure 6A), and the blue module was identified as having the highest correlation with CD8 T cells compared with other modules (cor = 0.9, p = 7.7e-63). The eigengenes and adjacencies were calculated according to their correlation (Figure 6B), and the five modules were divided into two main clusters. Furthermore, a heatmap was produced based on the interaction relationship of the six modules (Figure 6C). CD8 T cells recognize and kill cancer cells by expressing cytokines and cytotoxic molecules and, thus have been identified as a key target for immunotherapy. The blue module had the highest correlation with CD8 T cells, which suggested that the genes in blue module were candidates for immunotherapy biomarkers of sarcoma (Figure 6D, Supplementary Figure 7).
To further elucidate the function of the significant module, all genes in the blue module were analyzed with the “clusterProfiler” package in R to identify representative KEGG pathways and GO terms. As shown in Supplementary Figure 8, the most significantly enriched pathways of the blue module following KEGG pathway analysis were enhanced in chemokine signaling, the PI3K-AKT signaling pathway, and the JAK-STAT signaling pathway (Supplementary Table S2). GO enrichment analysis showed that the blue module contained biological processes mainly involved in chemokine-mediated, and complement receptor-mediated signaling pathways; and response to interferon-gamma, lipopolysaccharide, interferon-gamma, and chemokines. The cellular component (CC) was bent on the plasma, lysosomal, lytic vacuole membrane. Molecular function (MF) mainly enriched on the chemokine activity (Supplementary Table S2).
Identification and Validation of Hub Genes
Based on the following criteria (|MM| > 0.8, |GS| > 0.2, and the largest sub-network), four genes with high connectivity in the clinically significant modules were identified as hub genes (Figure 107). The “survival” package in R was performed to calculate the survival analysis (p < 0.05 as statistically significant). Table 2 shows that the survival analysis of hub genes identified CD48 antigen (CD48), putative P2Y purinoceptor 10 (P2RY10), and RAS protein activator like-3 (RASAL3). Figure 8 shows that these three genes were significantly and positively associated with overall survival. In addition, immunohistochemistry (IHC) staining data acquired from the HPAD also confirmed the differential expression of the predicted genes (CD48, P2RY10, RASAL3) in sarcoma samples (Supplementary Figure 9).
Genetic alterations
The CBioPortal database provides an open resource for exploring, visualizing, and analyzing multi-dimensional cancer genomics changes and clinical data. In this study, we used the CBioPortal database to estimate the genetic alterations in CD48, P2RY10, and RASAL3.Figure 9 shows that the frequency of mutations in CD48 was 11% in P2RY10 it was 9%, and in RASAL3 it was 12%. The three genes were altered in 22% (45/206) of the patients (Figure 9).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}