Experimental human participants. For cell line generation involving blood samples taken from human participants, informed consent, blood samples, and clinical evaluations were obtained under protocols approved by the Ethic Committee of Zhejiang University Schoolof Medicine and Institutional Review Board of Cincinnati Children’s Hospital Medical Center, which is in accordance with the Declaration of Helsinki. We confirm that all research was performed in accordance with relevant guidelines/regulations, and include in their manuscript a statement confirming that informed consent was obtained from all participants and/or their legal guardians.
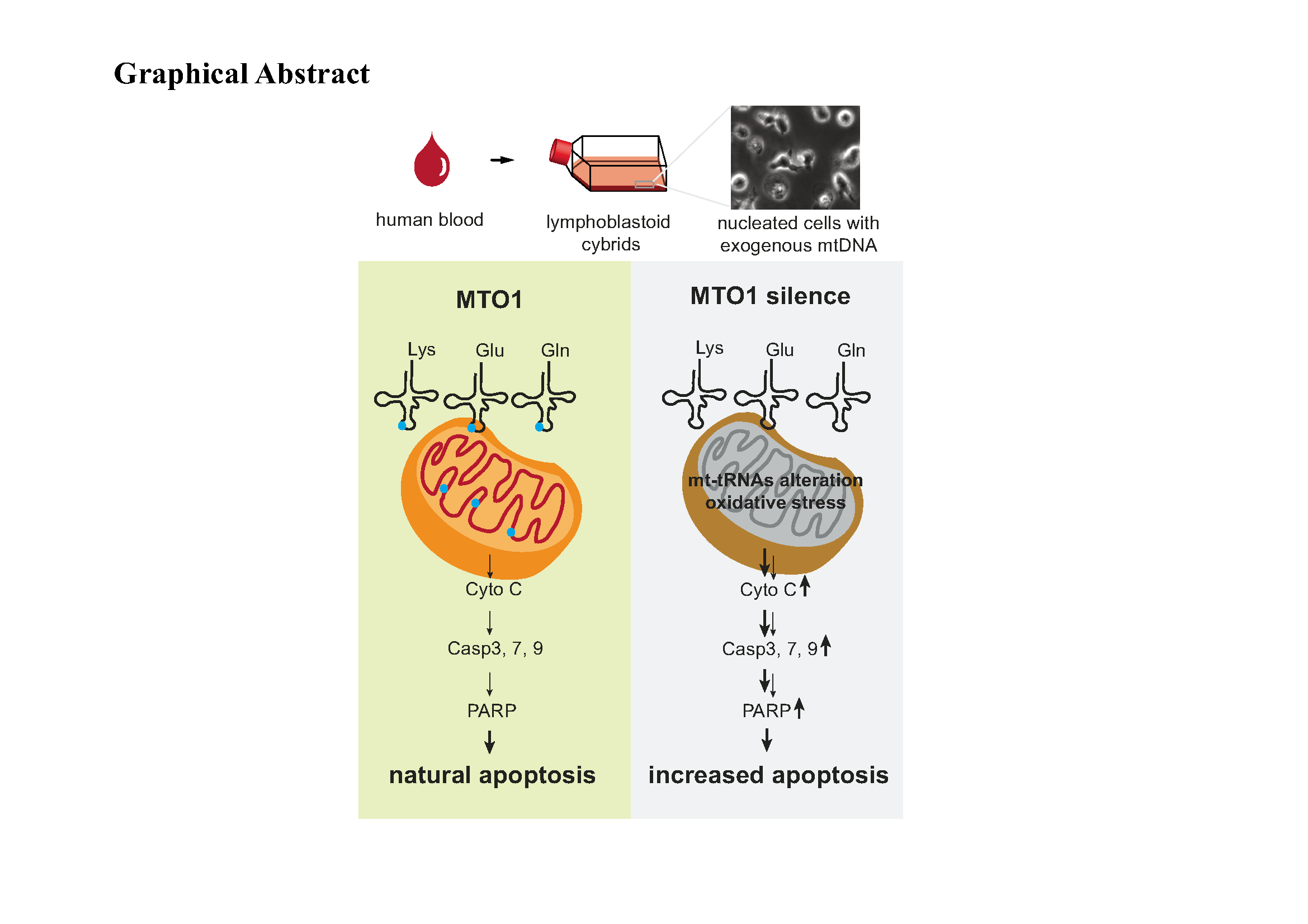
Generation of MTO1 knocking down cell lines. lymphoblastoid cell lines were derived from a healthy individual directly following previously described procedures [24]. In brief, cell lines were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS, Thermo Fisher Scientific). The mtDNA-less ρo206 cell line, derived from 143.TK- cell line was grown under the same conditions as the parental line, except for the addition of 50 ug of uridine/ml. Two cyrbid cell lines 143B-1 and 143B-2 were constructed with enucleated lymphoblastoid cell lines and were maintained in the same medium as the 143.TK- cell line.
The shRNA oligo primers targeting to silence MTO1 were:
Forward, 5’CCGGCCAGGGAGTTCAGCAAGATGACTCGAGTCATCTTGCTGAACTCCCTGGTTTTTG3’,
Reverse, 5’AATTCAAAAACCAGGGAGTTCAGCAAGATGACTCGAGTCATCTTGCTGAACTCCCTGG3’.
Lentiviral vectors, pLKO.1 with MTO1 shRNA or scramble shRNA, were co-transfected into HEK293T cells with psPAX2 and pMD2.G for lentivirus production. Complete medium was changed after 4-6 hours transfection. Virus was collected 48 hours after transfection and centrifuged at 12,000 g. Cell lines were cultured in medium containing virus for 48 hours. The stable shMTO1 and scramble clones were isolated by culturing cells in DMEM medium supplemented with 10% FBS and 1 µg/ml of puromycin for another 48 hours. The resultant clones were examined for the expression of MTO1 by Western blot analysis as detailed below.
Mitochondrial tRNA analysis. Total RNAs were generated using Totally RNATM kits (Ambion) from mitochondria isolated from knocking down and scramble control cell lines (∼2.0 × 108 cells), following manufacturer’s instructions. The levels of the thiouridine modification in the tRNAs were detected by the retardation of electrophoretic mobility in a polyacrylamide gel that contained 0.05 mg/ml ((N-acryloylamino)phenyl)mercuric chloride (APM) [20]. Ten µg of total RNAs were loaded in a 10% polyacrylamide-8 M urea gel electrophoresis in Tris–borate–EDTA buffer (TBE) and subsequently blotted onto positively charged membrane (Roche). To further discriminate unthiouridylated tRNA from thiouridylated tRNA, the samples were dethiolated through 1 hour incubation in EDTA-PBS buffer with H2O2 at room temperature and run in parallel. The membrane was incubated with specific non-radioactive DIG oligodeoxynucleoside probes at the 3′ termini according to the DIG Northern Starter Kit (Roche). Oligonucleotide probes for tRNALys, tRNAGlu, and tRNAGln were 5’-AAAGAGGTGTTGGTTCTCTTAATCTTTAAC-3’,
5’- ATTCTCGCACGGACTACAACCACGACCAAT-3’, 5’-AGGACTATGAGAATCGAACCCATCCCTGAG-3’, respectively. DIG-labeled oligodeoxynucleosides were generated by using the DIG oligonucleoside tailing kit (Roche). The bands were visualized by a CLINX ChemiScope bioimaging analyzer.
The aminoacylation assays was carried out as described elsewhere [25]. Briefly, total RNAs were isolated under acid conditions, and two µg of total RNAs was electrophoresed at 4°C through an acid (pH 5.2) 10% polyacrylamide–8 M urea gel to separate the uncharged and charged tRNA. The steady state tRNA Northern blot analysis was performed similar to thiouridine modification detection without APM. DIG-labeled oligodeoxynucleotide probes for tRNALeu(UUR), tRNATyr and 5S rRNA were: 5’-AGAAGAGGAATTGAACCTCTGACTGTAAAG-3’,
5’- GGTAAAAAGAGGCCTAACCCCTGTCTTTAG-3’,
5’- GGGTGGTATGGCCGTAGAC-3’, respectively. The hybridization and quantification of density in each band were performed according to the DIG Northern Starter Kit (Roche)
Western blot analysis. Western blotting analysis was performed as detailed previously [24]. Twenty micrograms of total proteins obtained from cells were denatured and loaded on sodium dodecyl sulfate (SDS) polyacrylamide gels. The gels were electroblotted onto a polyvinylidene difluoride (PVDF) membrane for hybridization. The antibodies used for this investigation were from Abcam [YARS2 (ab68725), ND3 (ab170681), and CO2 (ab110258), TRMU (ab50895), TOM20 (ab56783), GAPDH (ab8245), Cytochrome c (ab13575), total OXPHOS human WB antibody cocktail (ab110411)], Santa Cruz Biotechnology [ND4 (sc-20499-R), ND6 (sc-20667)), MTO1 (sc-398760)], Proteintech [TFAM (19998-1-AP), TFB2M (24411-1-AP), TUFM (26730-1-AP), KARS2 (14951-1-AP), LARS2 (17097-1-AP), CYTB (55090-1-AP), and ATP8 (26723-1-AP)], and Cell Signaling Technology [Caspase-3 (#9664), Caspase-7 (#8438), Caspase-9 (#7237) and PARP (#5625)]. Peroxidase Affini Pure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson) were used as a secondary antibody and protein signals were detected using the ECL system (CWBIO). Quantification of density in each band was performed as detailed previously [26].
Enzymatic Assays. The enzymatic activities of complex I, II, III, IV and V were measured as detailed elsewhere [27]. In brief, citrate synthase activity was analyzed by the reduction of 5,5′-dithiobis-2-nitrobenzoic acid at 412 nm in the assay buffer containing 0.1 mM DTNB, 50 µM acetyl coenzyme A, and 250 µM oxaloacetate. Complex I activity was determined with 10 µg/ml antimycin A and 2 mM KCN by following the decrease in the absorbance due to the NADH oxidation at 340 nm in assay buffer. The activity of complex II was analyzed by tracking the secondary reduction of 2,6-dichlorophenolindophenol (DCPIP) by DB at 600 nm in the assay buffer. Complex III activity was determined in the presence of 2 µg/ml antimycin A and 2 mM KCN by measuring the reduction of cytochrome c at 550 nm with reduced decylubiquinone in the assay buffer. Complex IV activity was measured by monitoring the oxidation of reduced cytochrome c as a decrease of absorbance at 550 nm in the assay buffer. Complex V activity was monitored by the oxidation of NADH at 340 nm in the assay buffer. All assays were performed by using Synergy H1 (Biotek, Winooski, VT, United States). Complex I-V activities were normalized by citrate synthase activity.
Measurements of oxygen consumption. The rates of oxygen consumption in mutant and wild type cell lines were assayed with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience), as detailed previously [24]. Non-mitochondrial OCR was analyzed as the OCR after rotenone/antimycinA treatment. Basal OCR was determined as OCR before oligomycin addition minus OCR after rotenone/antimycin A treatment. ATP-linked OCR was measured as OCR before minus after oligomycin addition. Proton leak was calculated as Basal OCR minus ATP-linked OCR. Maximal OCR was determined as the OCR after FCCP addition minus non-mitochondrial OCR. Reserve Capacity was defined as the difference between Maximal OCR minus Basal OCR. The protein content of each well was then measured to normalize OCR values.
ATP measurements. The Cell Titer-Glo® Luminescent Cell Viability Assay kit (Promega) was used for the measurement of cellular and mitochondrial ATP levels, following the modified manufacturer's instructions.
Assessment of mitochondrial membrane potential. Mitochondrial membrane potential was assessed with JC-10 Assay Kit - Flow cytometry (Abcam) according to general manufacturer's recommendations with some modifications as detailed elsewhere [24].
Flow cytometry for apoptosis analysis. For discrimination of apoptotic and non-apoptotic cells by Annexin V/ PI staining, cells were treated with or without 1 mM H2O2 (Sigma) for 4 hours. Cells were harvested and stained with Annexin V and 1 µl propidium iodide (PI) (V13242, ThemoFisher Scientific) according to the manufacturer’s instruction. Each sample was detected by FACSCalibur C6 (BD Biosciences) and analyzed using FlowJo software (Treestar).
Computer analysis. Statistical analysis was carried out using the unpaired, two-tailed Student’s t-test contained in the Microsoft-Excel 2013 or GraphPad Prism 7. Differences were considered significant at a p<0.05.
{kind=link}