Background Copy number variants (CNVs) are widespread among human genes, causing Mendelian or sporadic traits, or associating with complex diseases. Several tools have been developed for CNV assessment based on next generation sequencing (NGS) data using Read-depth (RD) strategy. However, maintaining high level of sensitivity and specificity is always challenging. Here, we present a novel, powerful, user-friendly and open accessed tool, T-CNV for CNV detection and visualization in targeted NGS panel.
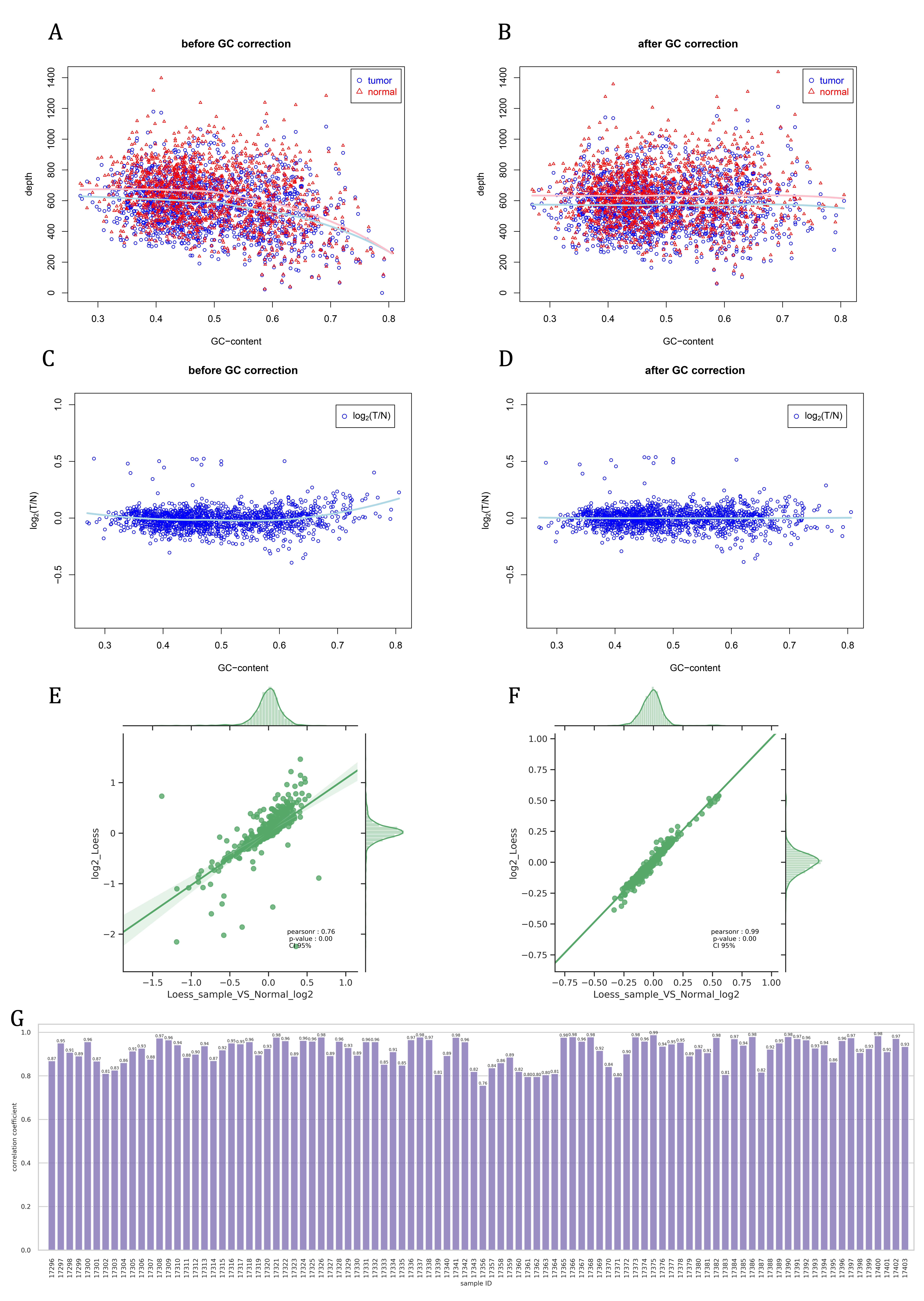
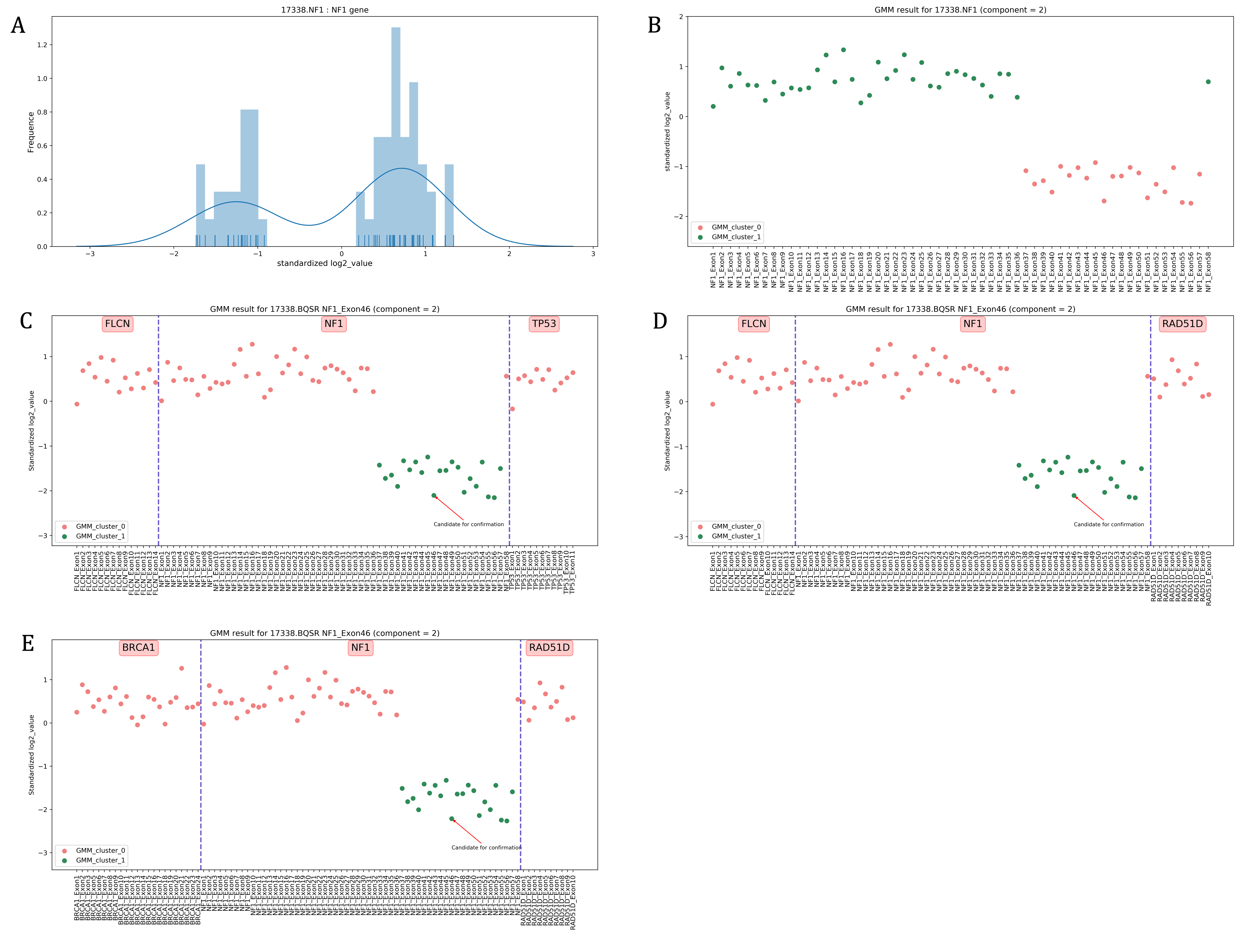
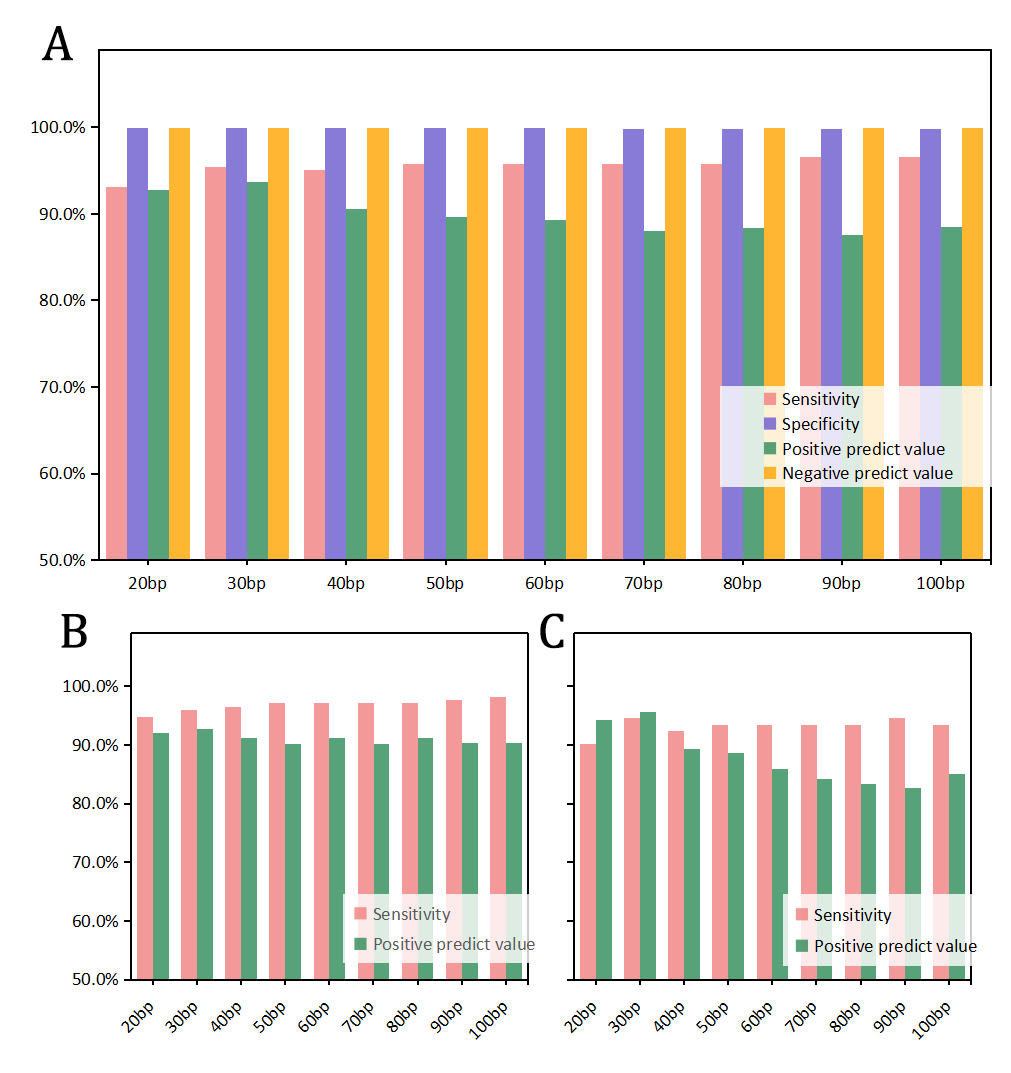
Results T-CNV consists of primary CNV detection and CNV candidates confirmation steps. After computing log2 values of normalized read depth ratio of tumor and normal/control sample, T-CNV confirms each possible CNV candidates by bins method, Gaussian Mixture Model (GMM) clustering approach and window-sliding method. We benchmarked its capacity with MLPA-validated dataset. Compared to three other advanced tools, T-CNV presents excellent performance with 95.42% sensitivity, 99.93% specificity and 93.63% positive predict value in MLPA-validated dataset, while achieving satisfactory performance in simulation study (sensitivity 65.95%, positive predict value 88.71% at coverage 100X).
Conclusions T-CNV is a novel and robust tool for CNV detection and visualization in targeted NGS panel consisting of determination of possible CNV candidates and further confirmation by three different methods. It’s publicly available at https://github.com/Top-Gene/T-CNV.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}