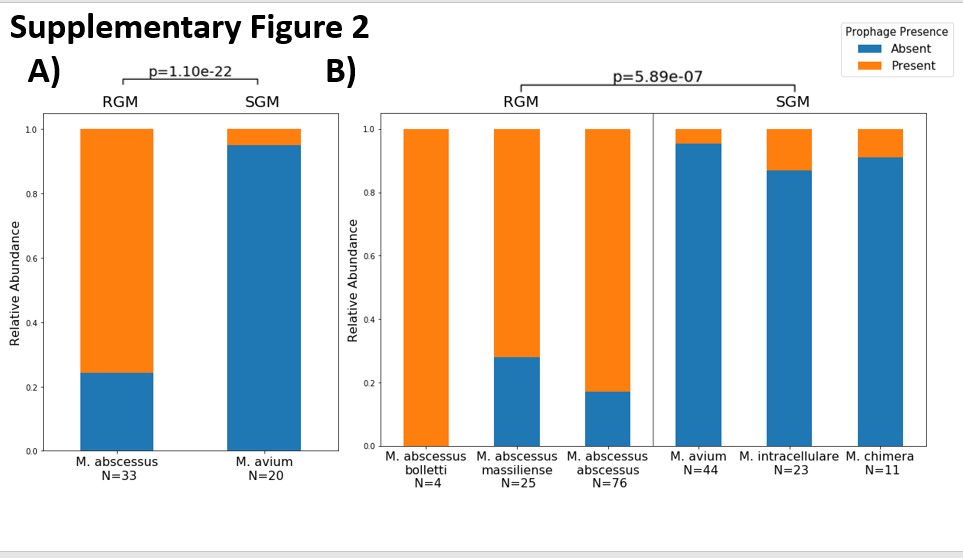
NTM infections can cause serious pulmonary disease that may become chronic and affect quality of life (1). Many of the mycobacterial species that cause NTM infections are ubiquitous in the environment and are known to thrive in built environments, including premise plumbing (2–4). The mechanism by which these organisms, which have long been recognized as environmental, become clinical pathogens is an active area of research with prior studies exploring host susceptibility (5), geographic factors (6), and genomic phylogeny (7). Mycobacterial species are categorized into two broad groups based on differing growth rates in culture; rapidly growing (RGM) and slowly growing (SGM). Of the six species/subspecies examined in this study M. avium, M. chimaera, and M. intracellulare are described as having a slow growth rate, requiring more than 7 days to see colonies in culture. M. abscessus subsp. massiliense, M. abscessus subsp. abscessus, and M. abscessus subsp. bolletii have a rapid growth rate, requiring less than 7 days to be visible in culture (8).
Bacteriophages are double stranded DNA viruses that infect bacteria and are known to transfer genetic material between bacteria through a process called transduction (9). Mycobacteriophages are bacteriophages that target mycobacteria. Environmental bacteria are subject to external pressures to adapt their genomes through horizontal gene transfer, including bacteriophage transduction (9). Bacteriophages can exhibit both lysogenic and lytic phases during a life cycle. The lysogenic phase involves a bacteriophage integrating genetic material into a bacterial genome and replicating in tandem until an external stimulus transitions the integrated bacteriophage, also known as a prophage, into a lytic life cycle. During the lytic phase, the bacteriophage utilizes the bacterial cellular machinery to create new phage particles that are then released during bacterial cell lysis. The newly created phage particles package bacterial genes at a low frequency, which are subsequently transduced during a new infection (10). Given the immense number of bacteriophage-bacterial interactions, transduction events are estimated to occur frequently in the environment (11).
Virulence is a general term that describes a pathogen’s invasive power, ability to overcome host defenses, and the replication efficiency of a pathogen within a host (12). Bacterial susceptibility to bacteriophages is positively correlated with the overall virulence of the bacteria (13). There is a selective advantage for bacteria that contain prophages with genetic elements capable of increasing fitness and propagative success. It is possible that genes carried by mycobacteriophages during transduction events could impact virulence as seen in other bacteria such as Vibro cholerae, Corynebacterium diphtheriae, and Streptococcus pyogenes (14). Prophages in these serious bacterial pathogens contain elements that contribute to quorum sensing, enzymatic functions, and even extracellular toxicity (14, 15).
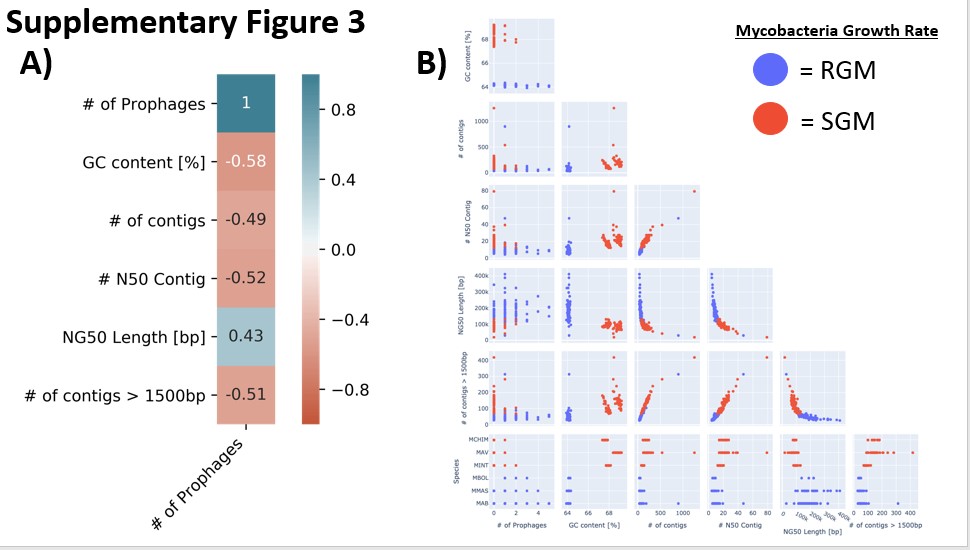
Here, we explore the frequency of integrated prophages in various NTM genomes and characterize the composition of bacterial genes within predicted prophage elements. Genetic elements including tRNAs act as a potential insertion site for mycobacteriophages (16). Our hypothesis is that more tRNAs are associated with an increase in the abundance of integrated prophages due to there being more targets for integration. CRISPR elements are a bacterial defense mechanism against viral integration, and CRISPR elements with fewer spacer elements are more susceptible to prophage integration (17). We hypothesize that the presence of CRISPR elements will reduce the number of integrated prophage elements.
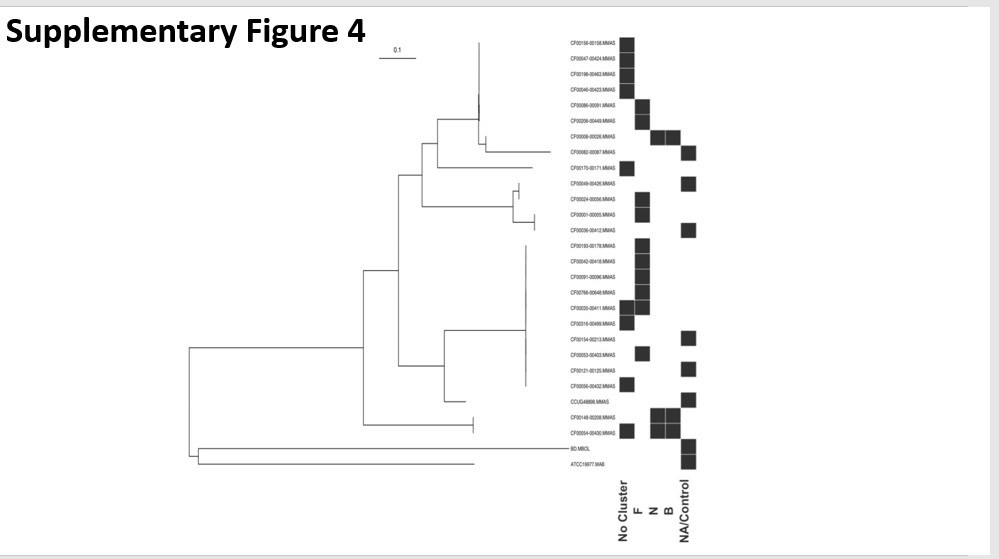
To explore differences between clinical prophages and environmental mycobacteriophages, we utilized PhageDB, which is a data repository of mycobacteriophages isolated from the environment (18). Most of these mycobacteriophages were identified, using a phage plaque screening assay to identify lytic mycobacteriophage capable of lysing the non-pathogenic species M. smegmatis. Mycobacteriophages from PhageDB are organized into sequence clusters, indicated by letters (A-Z), based on sequence similarity and shared functional protein families (19). Lettered clusters typically exhibit similar lifestyle and functional behavior. Prior works have suggested PhageDB mycobacteriophages clusters N, K, G, and A are capable of infecting clinical NTM (20, 21). Our hypothesis was that prophages from clinical genomes may be enriched for bacterial virulence genes compared to environmental mycobacteriophages from PhageDB. Exploring this hypothesis will elucidate the ability of prophages to act as a genetic repository for bacterial virulence genes within clinical NTM genome.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}