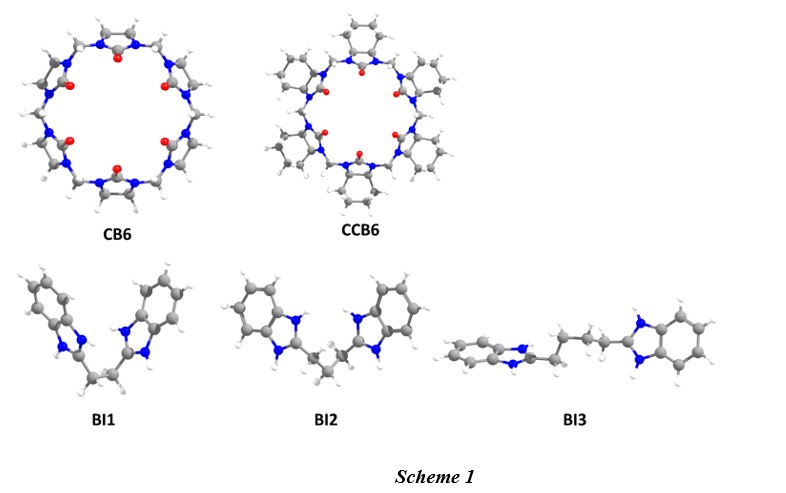
Structure of guest and host molecules
The optimized structures of cucurbit[6]uril (CB6) and cyclohexanocucurbit[6]uril (CCB6) are shown in Fig. 1a and b in lateral view and 1c and 1d in on top view respectively. The computed cavity diameter between the oxygen portals and the cavity depth of CB6 is 6.92 Å and 6.09 Å respectively with D6h symmetry. The observed values are in close agreement with the previously reported theoretical values [34]. The computed oxygen portals diameter in CCB6 is 6.77 Å and its cavity diameter is 5.96 Å, which are shorter than the values computed for the CB6 molecule [35]. Thus, the introduction of cyclohexanone groups in the glycouril part grades in the contraction of the oxygen portals and cavity diameter.
The studied three guest molecules are 1,ω-bisbenzimidazolyl derivates and have similar geometry with two benzimidazole connected by alkyl chain, however, the length of the alkyl chain various (Scheme 1). In the guest BI1, the two benzimidazole unit is connected by an ethylene moiety at the 2nd carbon of the heterocyclic ring. In guest BI2, propylene units connect the two benzimidazole groups, while guest BI3 has butane as a connecting unit. Due to the existence of a flexible interconnecting alkyl group, the guest molecules can adopt multiple configurations. Therefore, we have identified the lowest energy conformer for each of the studied guest molecules, which are shown in Fig. 1 (e-g). The other low-lying configurations for the guest molecules are shown in supporting information Figure S1. It is found that all the guest molecules were found to have a similar bent structure as the lowest energy conformer, while the leaner structures are high energy conformers.
Host-guest complexes between the studied CB6/CCB6 host and the guest molecules can exist in many possible conformers and with 1:1 and 2:1 stoichiometry [36, 37]. The present study is aimed at the complexation of the 1:1 stoichiometry and to understand the nature of interaction which helps for the most stable, rigid complex and exhibits high binding energy of formation between the guest and host molecules. The coordination of the guest with a host can exist in three binding modes (i) in which one of the guests aromatic rings are entrapped in the cavity (ii) in which the aliphatic chain is encapsulated and the bibenzylimidazole groups are on top and bottom of the CB6/CCB6 carbonyl portals (iii) the exclusion complex, in which the entire guest is outside the supramolecular cavity. The fully optimized structures of the lowest energy conformers in two orientations (on top and lateral view) for CB6 and CCB6 hosts with three of the guest molecules are shown in Fig. 2. The low-lying energy isomers for the CB6 complexes are shown in Figure S2 and CCB6 complexes in Figure S3. For all the studied complexes, the inclusion complexes were found to be more stable than the surface adsorption of guests on the host molecules. We have previously observed similar results in the complexation of cucurbiturils and pillararnes with various guest molecules. Recently, Liu and coworkers, deduce the X-ray structure of BI1@CCB6 and BI2@CCB6 and found partially encapsulated complexes to exists in 1:1 stoichiometry. Our theoretical result agrees well with these findings and provides confidence in our computational results [38, 39].
Upon encapsulation, we observed a high structural reorganization both in the host cucurbit[6]uril and cyclohexanocucurbit[6]uril and the guest molecules. In the guest BI1 and BI2, irrespective of the host, upon encapsulation resulted in the partial exclusion of the guest, with the hydrogen on heterocyclic nitrogen existing in bonding the carboxyl portal of CB6/CCB6. On the contrary, for the BI3 guest, the encapsulation occurs by the full inclusion with the benzimidazole moiety protruding out of the CB6/CCB6 cavity, and with the benzimidazole moiety lying perpendicular to each other. This indicates that the carbon chain length of the guest molecules plays a vital role in dictating the encapsulation of guest molecules inside the CB6/CCB6 guest molecules. It is worth pointing out that experimentally, the crystals for BI3@CCB6 were not obtained even after several attempts, while the 1H NMR spectral analysis has shown the existence of a fully encapsulated complex.
To apprehend the structural changes during the encapsulation, we computed the deformation energy of the host and guest molecules which are provided in Table 1. Furthermore, the deformation energy indicates the higher stabilization of encapsulated complexes [40]. The deformation energy is obtained from the difference in energy values of the host and guest in their optimized lowest energy state and the energy obtained on the geometry of host and guest after encapsulation. The deformation energy on the host was found to change with the guest molecule. Among the guests, BI2 shows the highest deformation on the host molecule with 5.13 kcal mol− 1 and 5.58 kcal mol− 1 on the CB6 and CCB6 molecule. Among the host, BI2 shows the least deformation on encapsulation with CB6, while BI3 shows the least deformation energy with CCB6. On the contrary, the highest deformation on guest molecule was found for CCB6 for BI2 guests followed by CB6 guests on BI2. It should be pointed out that upon encapsulation, the carbonyl oxygen portal diameter of CCB6 got contracted to 5.86 from 6.77 Å in one surface and got enlarged to 7.06 Å in the opposite surface in BI2 encapsulated complex. In the BI2@CB6 complex, the carbonyl oxygen portal contracted to 5.81 from 6.92 Å. This indicates that both the deformation of the guest and host ensued in a specific conformation serving to the formation of most stable complexes. In the case of BI3 complexes, the CCB6 host underwent more deformation than the CB6, while the guest molecule has maximum deformation with the CB6 host.
Table 1
Computed deformation energy, binding energy and theormodyanmical paprameters for the enapsulated complexes with guest molecules computed at a M06-2X-D3/6-311G(d,p) level with host (CB6) and CCB6 in the solution phase.
|
Complex
|
Deformation energy (in kcal/mol)
|
Binding energy (in kcal/mol)
|
Theormodyanmical paprameters (in kcal/mol)
|
|
Guest
|
Host
|
ΔG°
|
ΔH°
|
ΔS°
|
|
BI1@CB6
|
4.13
|
3.34
|
79.21
|
-61.45
|
-75.56
|
-47.34
|
|
BI2@CB6
|
2.05
|
5.13
|
83.07
|
-63.67
|
-79.27
|
-52.30
|
|
BI3@CB6
|
2.92
|
1.95
|
87.64
|
-69.68
|
-85.55
|
-53.24
|
|
BI1@CCB6
|
4.06
|
3.24
|
47.90
|
-29.34
|
-46.45
|
-57.36
|
|
BI2@CCB6
|
4.52
|
5.58
|
44.85
|
-28.56
|
-43.87
|
-51.35
|
|
BI3@CCB6
|
2.24
|
3.24
|
57.34
|
-37.07
|
-56.93
|
-66.59
|
A close examination of the structures indicates the existence of hydrogen bonding between the carbonyl oxygen atom and the hydrogen atoms of benzimidazole dication. The number of hydrogen bonding was found to vary with the complex. The number of hydrogen bonding with the guest BI3 was maximum than the partially encapsulated complexes with guest BI1 and BI2. In BI3 guest molecule, the intramolecular distance between the two benzimidazole acidic hydrogen atom attached to the nitrogen is at a distance of 6.88 Å away from each other, which matches with the cavity diameter of the CB6 than the CCB6 host. Besides the two benzimidazole units are attached to the linear more flexible butyl groups compared to the other studied guest molecules. Thus, in general, the more flexible guest BI3, make stable hydrogen bonding with the carbonyl portal of the CB6 unit to form a more stable BI3@CB6 complex.
Energetic analysis and stability of complexes.
To identify the stability of the complexes, we computed the binding energies of the stable complexes. The computed binding energy is provided in Table 1, which is obtained as the energy difference between the total energy of the encapsulated complex and the total energy of the host and guest molecules at their most stable state. The binding energies of the encapsulated complexes vary with both guest and host molecules. The binding energies are higher for the CB6 host compared to the CCB6. The binding energies for the guest molecules with the CB6 host vary from 79.21 to 87.64 kcal mol− 1. In the case of CCB6, the observed values were in the range of 44.85 to 57.34 kcal mol− 1. Furthermore, the highest binding energy is observed for the BI3 guest molecule with CB6 host molecules. The higher binding energy of BI3 with CB6 can be attributed to the bond length of linear flexible butyl groups whose length matches with the CB6 cavity. Our calculated energy difference between the lowest energy BI3@CCB6 complex and the next higher energy isomer is just 9.35 kcal/mol, which implies that the two conformers can coexist during the complexation process, which eventually interrupts the crystallization of one isomer.
To deduce the complexation ability of these guest with the hosts CB6 and CCB6, we have computed the thermodynamic parameters viz., standard free energy changes (ΔG°), enthalpy changes (ΔH°) and the entropy (ΔS°) changes for the formation of these encapsulated complexes. Table 1 Summarizes the thermodynamic parameters for the encapsulated complexes. computed at 1 atm and 298.15 K using the partition functions derived from the vibrational frequency calculations. The enthalpy changes for all the complexes are all negative indicating the encapsulation process is exothermic. Furthermore, the Gibbs free energy change was also negative indicating the encapsulation process is spontaneous and thermodynamically favorable. The negative ΔG° and ΔH° values imply that the formation of the encapsulated complex is a spontaneous and enthalpy driven process [41]. The computed entropies were also negative implying that the system moves to a more ordered encapsulated system from the free fluctuating guest molecules along with the structural modification of the cucurbituril guests. It is interesting to observe that the ΔG° and ΔH° are larger for the CB6 encapsulated systems than the CCB6 complexes. Besides, in the CCB6 encapsulated complexes the BI3 complex has higher ΔG°, ΔH° and ΔS° suggesting that among the CCB6 complexes its formation would be more thermodynamically favorable, spontaneous with high orderliness. Thus, from the thermodynamic consideration, the formation of CB6 encapsulated complex formation is more facile, stable, and spontaneous compared to the CCB6 complexes.
The frontier molecular orbitals (FMO) of a complex helps in determining its chemical stability [42]. The Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) decides the materials' ability to donate and accept an electron during a chemical reaction respectively. The FMO of the inclusion complex is depicted in supporting information Figure S4. It can be seen from the figure, for all the encapsulated complexes the HOMO or LUMO is located mainly on the guest molecule principally on the benzimidazole moiety. This implies the electron density transfer happens mainly in the charged guest molecule and when excitation happens electron transfer happens between the locally excited states.
Besides the HOMO-LUMO gaps is an important factor that dictates the chemical stability of a system [43]. The higher the HOMO-LUMO gaps, the higher would be the stability of the system. The HOMO-LUMO gaps of the free guests, host molecules, and their inclusion complexes are provided in Table 2. The complexes were found to have HOMO-LUMO gaps comparable to that of free guest molecules, implying no appreciable change in their electronic properties upon their complex formation. Furthermore, the more stable complexes with BI3 guests were found to have the highest HOMO-LUMO gaps confirming their better stability computed through the binding energy calculation.
Table 2
Calculated HOMO-LUMO gap (Egap) and the reactive discreptors for the inclusion complexes chemical potential, electrophilicity indices, chemical hardness for the inclusion complexes of CB6 and CCB6 with BI1, BI2, and BI3 guest molecules computed in the solution phase at the M06-2X-D3/6-311G(d,p)-level of theory.
|
System
|
Egap (in eV)
|
µ (in eV)
|
η (in eV)
|
ω (in eV)
|
ECT
|
|
CB6
|
9.75
|
-3.51
|
4.88
|
1.26
|
-
|
|
CCB6
|
9.65
|
-3.42
|
4.80
|
1.22
|
-
|
|
BI1
|
7.83
|
-4.72
|
3.92
|
2.84
|
-
|
|
BI2
|
7.90
|
-4.63
|
3.95
|
2.71
|
-
|
|
BI3
|
7.95
|
-4.57
|
3.97
|
2.63
|
-
|
|
BI1@CB6
|
7.68
|
-4.54
|
3.84
|
2.68
|
0.49
|
|
BI2@CB6
|
7.80
|
-4.45
|
3.90
|
2.54
|
0.45
|
|
BI3@CB6
|
7.90
|
-4.28
|
3.95
|
2.32
|
0.43
|
|
BI1@CCB6
|
7.63
|
-4.49
|
3.82
|
2.65
|
0.49
|
|
BI2@CCB6
|
7.73
|
-4.43
|
3.87
|
2.54
|
0.46
|
|
BI3@CCB6
|
7.86
|
-4.32
|
3.93
|
2.37
|
0.44
|
The eigenvalues of FMOs can be used to compute the global chemical reactivity descriptors such as electronic chemical potential (µ), chemical hardness (η), global electrophilicity index (ω), and electrophilicity based charge transfer (ECT) [44]. The details to arrive at the descriptors are provided in our previous publications [45], and the values are listed in Table 2. The chemical potentials of all the complexes were negative, which implies that the encapsulated complexes are stable. Besides, the chemical potentials of free guest molecules are greater than the free host molecules which suggests that charge transfer can occur from the guest to the host molecule upon encapsulation. The chemical hardness of the guest molecules is higher than the host molecules and between the host molecules, no appreciable change in hardness values was observed. The higher the chemical hardness, the less probable would be the system to undergo electron transfer. The electrophilicity index ω, is a positive quantity and higher values are characteristics of most electrophilic systems. From the table, we can see the guest molecules due to their dicationic nature have higher ω values than the host molecules. Furthermore, among the guests, CB6 is a better electrophile than CCB6. Among the encapsulated complexes CCB6 complexes have a lower electrophilicity index ω, suggesting them to be a better host system for neutral guests compared to CB6 host.
To verify the direction of charge transfer we have computed the electrophilicity based charge transfer (ECT) using the Eq. (5)
ECT = (ΔNmax)host – (ΔNmax)guest (5)
where ΔNmax = µ/η
The obtained results for the encapsulated complexes are provided in Table 2. The ECT values for all the complexes were all positive, indicating the charge transfer occurs from the guest to host molecule, due to the dicationic nature of the guest molecule. To comprehend the involvement of charge transfer, electron density difference maps for the encapsulated complexes are plotted, which are depicted in the supporting information Figure S2 for CB6 and CCB6 complexes. Red areas represent the increased density and blue area represents the decreased density in the complex. In all the complexes, the carbonyl oxygen atom of CB6 and CCB6 experience an increase in electron density, while the hydrogen atom on benzimidazole chromone a decrease in electron density is observed. This build-up of charge around the carbonyl portal is more pronounced in the fully encapsulated BI3 complexes, which accounts for the higher interaction energy in those complexes.
Nature of Intermolecular Interaction
To understand the type of the main intermolecular interaction that stabilizes the encapsulated complexes and to label their nature and strength, we used quantitative molecular electrostatic potential analysis (MESP), Atoms-in-molecules (AIM) analysis, NCI-RDG, and EDA analyses. The obtained results are presented below.
Molecular electrostatic potential
Molecular electrostatic potential analysis (MESP) is generally used to understand the effective localization of electron density, especially in the supramolecular complexes where a charge transfer occurs from the guest to host or host to guest molecule [43, 45]. The MESP mapped isosurface for all the encapsulated complexes is provided in Fig. 3 and for the individual molecules are provided in Figure S5. The electron-rich regions are represented in red color and the electron-deficient region is depicted by the blue color. In the cucurbit[6]uril the electron-rich regions are found on the carbonyl portal and the electron-deficient regions are found in the cavity. The guest molecules which are dicationic are electron-deficient which are clearly envisaged from the blue color of the MESP plot. Upon complexation, we observe that most of the regions turn to blue shade, which indicates a high charge transfer from the guest to the host molecule. Among the complexes, a high electron density transfer is observed for the BI3@CB6 and BI3@CCB6 complexes, from the carbonyl portal to the BI3 guest molecule. This accounts for the higher stability of those complexes compared to the other encapsulated complexes.
To corroborate the above information we have calculated the quantitative molecular electrostatic potential values for the free guest, host, and the encapsulated complexes at 0.001 electrons/Bohr3 isodensity surface [46]. The observed positive and negative potentials which are designated as Vs,max and Vs,min respectively were all positive for the guest molecule due to the dicationic in nature. In the host CB6, the Vs,max value of 52.31 kcal/mol was observed at the middle of the eight-membered ring, and two negative Vs,max with values of -29.51 and − 42.09 kcal/mol were observed inside the cavity one close to the five-membered glycoluril unit and at the middle of the eight-membered ring respectively. The Vs,min value of -111.76 kcal/mol was observed on top of the carbonyl portal. In the case of the CCB6 molecule, we observed a positive Vs,min of 26.06 kcal/mol on the cyclohexanone methylene group and a negative value of -129.56 kcal/mol on the carbonyl portal. In the CCB6 host, we observed several positive Vs,max values on the cyclohexane methylene hydrogen atoms with values of 44.98 kcal/mol and with two negative Vs,max with values of -60.76 and − 48.65 kcal/mol were observed inside the cavity one close to the five-membered glycoluril unit and at the middle of the eight-membered ring respectively. The close observation of quantitative molecular electrostatic potentials in the host’s molecule shows the presence of higher negative potential on the carbonyl portal and positive Vs,max values on CCB6 host compared to the CB6 molecule.
Upon encapsulation, we observe the Vs,max values on both the host CB6 and CCB6 turned to positive values in most cases. The Vs,min of partially encapsulated complexes with guest BI1 and BI2 have negative values on the carbonyl portal opposite to the encapsulated parts, however, the values are considerably lower than their bare host molecules. The Vs,min values on the carbonyl portal near the encapsulated regions were found to have a high positive value, while the charge on the benzimidazole hydrogen has considerably got reduced. A comparison between the BI3 and other guest encapsulated system, show that in BI3 both the Vs,max and Vs,min turn to positive values. These results suggest that the distribution between the host and BI3 molecule is more pronounced. Furthermore the Vs,max value on the benzimidazole hydrogen has got reduced to 62.85 kcal/mol from 108.55 kcal/mol, implying the acidic hydrogen attracts the charge density from the host molecule. Vs,max values on the methylene group which acts as a spacer between benzimidazole group has gained only less positive charged after encapsulation. Thus, from the above results, we can conclude that in BI3, the spacer length has helped for the charge transfer from the two benzimidazole groups compared to the other guest molecules.
AIM analysis
The atoms in molecules (AIM) has been used to analyze the nature of interactions in many molecular systems and to categorize bonding interactions in terms of a quantum mechanical parameter the electron density [47, 48]. The quantity of electron density at the bond critical point (BCP) provides information about the strength of bonding between the two molecules in any supramolecular complexes. The computed total electron densities at the BCP along with the total intermolecular BCP number are provided in Table 3 for all the encapsulated complexes. The molecular graphs obtained for all the encapsulated complexes are shown in Fig. 4. It is evident that several of these BCPs have total electron density are in the range between 0.01 to 0.04 a.u. which can be classified as hydrogen bonding. It is interesting to find that the total number of intermolecular BCP’s and total electron density at the BCP’s in the encapsulated complex varies by guest and host. The computed electron density per BCP’s is found to be higher for BI3 guest which agrees well with their higher computed binding energy calculations.
Table 3
QTAIM summation parameters corresponding to intermolecular bonding between the guest BI1, BI2 and BI3 and CB6/CCB6 host molecule in the host-guest complexes computed at the M062X-D3/6-311G(d,p) level of theory. The provided values are in atomic units.
|
Complexes
|
Σ(ρ)ρ
|
Σλ1
|
Σλ2
|
Σλ3
|
Σ∇2ρ(ρ)
|
ΣG(ρ)
|
ΣV(ρ)
|
ΣΗ(ρ)
|
Average
- ΣG(ρ)/ ΣV(ρ)
|
Number of BCP’s
|
|
BI1@CB6
|
0.249427
|
-0.241048
|
-0.197211
|
1.349685
|
0.911427
|
0.199961
|
-0.172066
|
-0.027896
|
1.162118
|
23
|
|
BI2@CB6
|
0.301850
|
-0.306907
|
-0.258491
|
1.668798
|
1.103399
|
0.244910
|
-0.213970
|
-0.030940
|
1.144600
|
27
|
|
BI3@CB6
|
0.264485
|
-0.272668
|
-0.200436
|
1.493464
|
1.020359
|
0.222045
|
-0.189000
|
-0.033045
|
1.174841
|
23
|
|
BI1@CCB6
|
0.239112
|
-0.220356
|
-0.171598
|
1.261291
|
0.869338
|
0.189813
|
-0.162291
|
-0.027522
|
1.169584
|
23
|
|
BI2@CCB6
|
0.242413
|
-0.222664
|
-0.180765
|
1.263100
|
0.859671
|
0.186768
|
-0.158617
|
-0.028150
|
1.177478
|
25
|
|
BI3@CCB6
|
0.272185
|
-0.285365
|
-0.219852
|
1.568082
|
1.062864
|
0.233698
|
-0.201680
|
-0.032018
|
1.158756
|
22
|
The sign of Laplacian is determined by energy that dominates in the bonding zone. A positive ∇2ρ indicates that kinetic electron energy density G(ρ) is greater than the potential electron energy density V(ρ). A positive ∇2ρ is observed in closed-shell systems/noncovalent interaction and is indicative of the depletion of electronic charge between the nuclei, and a negative value indicates a covalent bond [49, 50]. More basic information on the AIM can be found in previous publications [34, 35, 38, 39]. Furthermore a positive value of H(ρ), also indicates the existence of noncovalent interaction at the BCP. Besides the above terms, if the ratio of -G(ρ)/V(ρ) > 1, then the interaction can also be classified as noncovalent [51]. The observed topological parameters namely ∇2ρ, G(ρ), V(ρ), and H(ρ) at the intermolecular BCP’s of all the complexes are provided in the supporting information Tables S1-S6 and their summation values are provided in Table 3. We observe the presence of C···O, C···N, N···O, C···H, H···N type of interaction in addition to the conventional C-H····O = C hydrogen bonding interactions. In the case of BI3 guest, we observe more number of C-H····O = C BCPs than the other nonconventional intermolecular interactions. Furthermore, the ρ values are higher for the C-H····O = C interaction between the benzimidazole hydrogen and the carbonyl portal of the guest molecule, especially in the case of the BI3 guest molecule.
Rozas and coworkers, classified the interactions based on ∇2ρ and H(ρ) values as (i) strong H-bonds with covalent character if the values of ∇2ρ < 0 and H(ρ) < 0. (ii) medium-H bonds with partial covalent character, if the values of ∇2ρ > 0 and H(ρ) < 0 and (iii) weak H-bonds with major electrostatic character, if the values of ∇2ρ > 0 and H(ρ) > 0 [52, 53]. From the topological parameters in Table S1 – S6, the values of H(ρ) were found to be all positive and ∇2ρ were all small positive values implying the existence of weak interaction, mainly of electrostatic in nature. Besides the ratio of G(ρ)/V(ρ) is > 1 for all the complexes, supporting the existence of weak intramolecular bonding.
Noncovalent Interaction Analysis – Reduced Density Gradient (NCI-RDG) analysis
The method of noncovalent interaction analysis (NCI) provides the graphical visualization of the regions where non-covalent interactions occur in real-space and was demonstrated to be capable of distinguishing hydrogen bonds, van der Waals interactions, and repulsive steric interactions [54, 55]. Recently, we have shown that NCI-RDG analyses can recognize weak interaction definitely than the AIM analysis [56]. To know the existence of weak interactions, NCI plots are generated with the plot of the RDG S versus (sign λ2)ρ, where (sign λ2)ρ is the electron density multiplied by the sign of the second Hessian eigenvalue (λ2). The value of (sign λ2)ρ is helpful to predict the nature of interaction; for a repulsive interaction (sign λ2)ρ > 0 and attractive interaction (sign λ2)ρ < 0 interactions.
The NCI plots for all the CB6 encapsulated complexes are shown in Fig. 5, along with the 2D RDG plots, while the NCI plots for CCB6 encapsulated complexes are depicted in Figure S6. In the 2D scatter plots the stronger interactions which are shown in blue color are seen at the higher density values (ρ > 0.01 a.u.), and the weaker interaction which is shown in green color including that of dispersion will show a spike in regions with ρ < 0.01 a.u. The steric repulsion which is depicted with red color is observed above ρ < 0.02 a.u. We observe broad spikes in the high ρ regions 0.03–0.04 a.u. which arises from the hydrogen bonding. In the encapsulated complexes with BI1 and BI2 as guest the strong interaction show spikes in the region − 0.03 a.u., while in BI3 encapsulated CB6 and CCB6 complexes, we observe a broad spike due to the existence of multiple hydrogen bonding in the systems. Equally, some low-gradient spikes were observed near − 0.02 a.u. which are due to the existence of hydrogen bonding interaction between the guest molecules methylene hydrogen’s and the host's carbonyl oxygen atoms. Also, we observe spikes due to the van der Waals forces near − 0.01 a.u. in all the complexes. This spike due to van der Waals forces are broader in the BI1 and BI2 guest encapsulated complexes, exemplifying the dominant role of weak van der Waals forces than the BI3 guest encapsulated complexes.
The 3D spatial isosurface visualization of NCI for all the CB6 encapsulated complexes is shown in Fig. 5 and for CCB6 it is shown in Figure S4. In the above figures, the van der Waals forces are shown in green patches, steric hindrance in red patches, and hydrogen bonding with blue patches. In all the complexes, we observe the existence of splattered green patches especially in the regions with eight-membered cyclic rings of CB6 and CCB6 units. The presence of such splattered green patches has been attributed to the charge perturbation which occurs due to the electrostatic interactions between the charged guest and neutral host molecules. Besides that, pronounced red patches are observed in the five-membered ring due to the increased steric hindrance in the encapsulated complexes. In the BI1@CB6 and BI1@CCB6 complexes, we observe the presence of a partially blue patch due to the interaction between the benzimidazole hydrogen atom and the carbonyl portal of CB6 and CCB6 host molecules. In the BI2@CB6 and BI2@CCB6 complexes, the above interaction was found to be more pronounced and see as an intense blue patch and this happens on one side of the carbonyl portal. In the case BI3@CB6 and BI3@CCB6 complexes, two such intense blue patches were observed on each side of the portal originating from the two hydrogen atoms present on the cationic benzimidazole nitrogen atoms. Thus, in the BI3@CB6 and BI3@CCB6 complexes, the higher stability can be attributed to the more pronounced hydrogen bonding between the hydrogen atoms of the cationic benzimidazole unit and the portal oxygen atom of the carbonyl group in the host molecules.
EDA analysis
To quantitatively know the strength of intermolecular interactions in the encapsulated complexes between the guest and host molecule, we used the energy decomposition analysis (EDA). In EDA analysis the total interaction energies were decomposed into Pauli repulsion, electrostatic, orbital and dispersion energy terms using the Ziegler-Rauk decomposition analysis as is evaluated using Eq. 6 [57].
ΔEint = ΔEPauli + ΔEelst + ΔEorbital + ΔEdisp (6)
where ΔEPauli is Pauli’s repulsion energy, ΔEelst, ΔEorbital, ΔEdisp is electrostatic interaction, orbital and dispersion energies. ΔEPauli is always positive due to the destabilizing interactions associated with proper antisymmetrization and renormalization of the Hartree product of the fragment wavefunctions and is accountable for steric repulsion. ΔEorbital is stabilizing component originating from the interactions of occupied molecular orbitals of one fragment and the unoccupied molecular orbitals of the other fragment, or the mixing of occupied and virtual orbitals. ΔVelst corresponds to the classical electrostatic interaction between unperturbed charge distributions between the fragments.
The calculated EDA values along with the percentage of contribution for the encapsulated complexes are provided in Table 4. Pauli’s repulsive energy for all the complexes is similar, except the BI3@CCB6 complex. The high BI3@CCB6 Pauli’s value for the complex indicates the higher involvement of destabilizing factor involved. In stabilization terms, the largest contribution is attributed to the electrostatic energy presumably due to the dicationic charge of the guest molecules. Also, it can be seen that the orbital and dispersion terms have an almost equal contribution. Among the complexes, the BI3@CCB6 has the highest % contribution of 55 percent and among the guest molecules, BI3 shows higher energy towards electrostatic attraction energy. ΔEorbital terms which account for the covalent nature associated with a charge transfer between the fragments were found to have a contribution in the range of 23.62 to 26.21 % towards interaction energy, which supports the charge transfer occurs mainly within the guest/host molecule. This observed result supports the result observed in the FMO and ECT study. The ΔEdisp has a contribution in the range of 20.27 to 23.27 %. It is interesting to observe that those complexes with high ΔVelst have low ΔEdisp values. Thus, from the EDA results, we can conclude that noncovalent and electrostatic interaction with partial covalent character stabilizes the encapsulated complexes.
Table 4
Energy decomposition analysis for the encapsulated complexes at the B3LYP-D3/TZ2P level of theory on the optimized geometry.
|
Complexes
|
∆Epauli
|
∆Eelst
|
∆Eorbt
|
∆Edis
|
|
BI1@CB6
|
81.12
|
-117.25 (51.28)
|
-58.24 (25.47)
|
-53.12 (23.23)
|
|
BI2@CB6
|
82.46
|
-117.70 (50.98)
|
-60.52 (26.21)
|
-52.62 (22.79)
|
|
BI3@CB6
|
88.20
|
-134.16 (54.57)
|
-58.07 (23.62)
|
-53.60 (21.80)
|
|
BI1@CCB6
|
81.44
|
-125.21 (52.28)
|
-58.56 (24.45)
|
-55.72 (23.26)
|
|
BI2@CCB6
|
75.15
|
-117.69 (52.09)
|
-55.82 (24.70)
|
-52.44 (23.21)
|
|
BI3@CCB6
|
94.23
|
-150.39 (55.33)
|
-66.30 (24.39)
|
-55.09 (20.27)
|
{kind=link}
{kind=link}