Mosquito species
The non-infected Aedes albopictus strains used in the present study were maintained in the bio-secure insectary of the Insect Pest Control Laboratory (IPCL), Joint FAO/IAEA Division of Nuclear Techniques in Food and Agriculture, Seibersdorf, Austria and reared following the FAO/IAEA guidelines [22]. In brief, mosquito strains were kept under standard laboratory conditions at a temperature of 27 ± 1 °C, 60 ± 10% relative humidity, and a photoperiod of 12:12 (L:D) h including dusk (1 h) and dawn (1 h) transitional periods [22, 23]. Adults were kept in standard 30 ´ 30 ´ 30 cm Bugdorm cages (Megaview Science Education Services Co. Ltd., Taichung, Taiwan) in an insectary deprived of natural light and continuously supplied with 10% wt:vol sucrose solution. Before total RNA extraction, adults were starved for 12 h to empty stomach content and stored at -80 °C.
Virus-infected mosquito samples
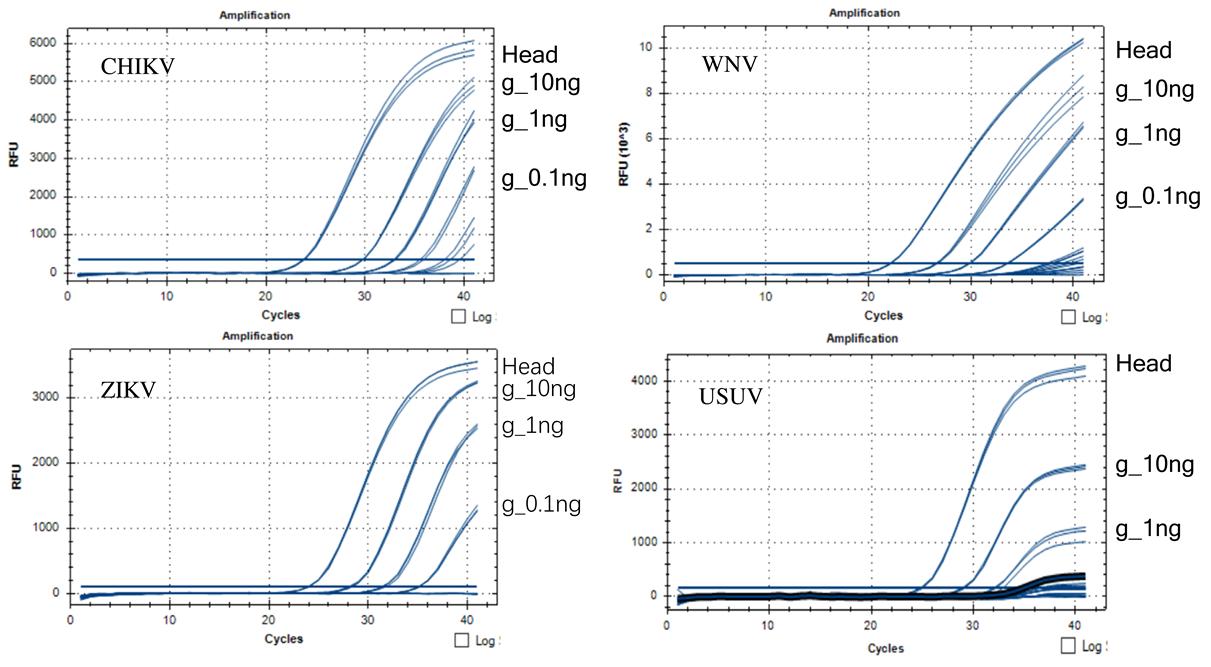
The project ‘Research infrastructures for the control of vector-borne diseases’ (lnfravec2, https://infravec2.eu/) provided the BORA strain of Ae. aegypti, infected with CHIKV and ZIKVs, and the Gavà strain of Culex pipiens, infected with WNV and USUV. In brief, 5–7-day old females of the Ae. aegypti BORA strain were infected with CHIKV and ZIKV by feeding them on an infectious blood meal with virus titers of 1.5 ´ 107and 1.02 ´ 107 plaque-forming units (PFU)/ml respectively. The virus titration was performed by the plaque assay for CHIKV and ZIKV and expressed in PFU/ml as previously described [24, 25]. A Hemotek® system (Discovery Workshops, Accrington, UK) was used for feeding the adult females and engorged females were fed with 10% sucrose in a chamber incubated at 28 °C and 80% humidity for 14 days in the bio-secure insectary of the Maladies Infectieuses et Vecteurs: Ecologie, Génétique, Evolution et Contrôle, Institut de Recherche pour le Développement (IRD), Montpellier, France. In addition, Cx. pipiens females inoculated with WNV and USUV were prepared at the Centre de Recerca en Sanitat Animal (Campus of Autonomous University of Barcelona) Barcelona, Spain. Culex pipiens females were inoculated intrathoracically with 1–2 µl per adult using a stock of WNV (7.52 log10 TCID50/ml) and USUV (6.88 log10 TCID50/ml). Virus titers were determined by a standard limiting dilution assay [26] using monolayers of cells employed for virus propagation. Viral titers were expressed in 50% tissue culture infectious dose (TCID50) per ml. Injected females were kept at 28 °C and 80% humidity for 7 days. Bodies and heads were separated in individual tubes and homogenized in 500 µl of TRIzol (Invitrogen, Carlsbad, California, USA) and kept at -80 °C. Infected heads and bodies were delivered to the IPCL for RNA extraction and further analysis. The level of infection of infected mosquito bodies was estimated by evaluating the virus infection titer in the corresponding head using quantitative RT-qPCR. Only bodies of which the corresponding head showed high virus titer were considered infected and were used in spiking the non-infected mosquito samples.
Spiking non-infected mosquito samples with virus-infected mosquitos
To determine the ability of the quantitative RT-qPCR assays to detect MBVs, a single Ae. aegypti infected with CHIKV or ZIKV and Cx. pipiens infected with WNV or USUV, was homogenized within large pools of uninfected adult Ae. albopictus mosquitoes, i.e. n = 165 (100 mg), n = 320 (200 mg) and n = 1600 (1000 mg) and total RNA extracted. A pool consisting of only uninfected mosquitoes was included as a negative control. The negative control, and the 320 and 1600 mosquito pools were replicated twice.
Total RNA extraction
Total RNA was extracted from mosquitoes using TRIzol Reagent (Invitrogen, Carlsbad, California, USA) according to the supplier’s instructions. Based on the size of the pool, adult mosquitoes were homogenized either in microtubes with a sterile pestle or with a sterilised mortar and pestle after adding liquid nitrogen. After grinding and homogenizing mosquito adult pools using liquid nitrogen, TRIzol reagent containing one single infected mosquito was added to the uninfected mosquito pool and the RNA extraction was carried out. For the negative control pool, TRIzol reagent was added after grinding with liquid nitrogen. The total RNA pellet was resuspended in 100, 200 and 1000 µl of RNase-free water for the pools composed of 165, 320 and 1600 mosquitoes, respectively. The quantity and quality of RNA samples were determined using a Synergy™ H1 microplate reader (BioTek, Winooski, Vermont, USA). The RNA samples were serially diluted in ten-fold steps from 10 to 0.0001 ng/µl for a concentration that will consistently give the same amount per well in the RT-qPCR and were stored at -80 °C.
Evaluation of primers and probes specificity
To investigate the possibility of using multiple primers and probes sets to detect several viruses in the same reaction, we tested the specificity of each primer and probe set to detect other MBVs. The total RNA extracted from uninfected mosquitoes was tested to ensure that there were no false positives caused by cross-reactions with the host-species. This step also determined any background signal generated by primer cross-reactivity with mosquito-derived RNA.
Generation of standard curves for the RT-qPCR
To obtain a stable positive control for the detection of MBVs, the RNA sequences containing and flanking the sequence regions of the virus specific primers and probes of CHIKV, ZIKV, WNV and USUV were amplified using the primers listed in Table 1 and cloned into pGEM-T vector (Promega, Madison, Wisconsin, USA). The primers in Table 1 were selected using the primer3 software with the default setting (http://bioinfo.ut.ee/primer3-0.4.0/) and the sequence of CHIKV (GenBank: NC004162), USUV (GenBank: AY453412), ZIKV (GenBank: AY632535) and WNV (GenBank: DQ211652). The SuperScript® III First-Strand Synthesis System for RT-qPCR (Invitrogen) was used to synthesize first-strand cDNA from purified poly(A)+ or total RNA following the manufacturer’s instructions. The targeted sequences were amplified by Taq PCR Master Mix (Qiagen) with the following PCR conditions: 5 min at 94 °C; 35 cycles of 30 s at 94 °C, 30 s at 58 °C and 1 min at 72 °C; and 10 min at 72 °C. The PCR product was purified using the High Pure PCR Clean-up Micro Kit (Roche, Basel, Switzerland) and ligated to the pGEM-T vector (Promega, Madison, Wisconsin, USA), following the supplier’s instructions. The recombinant plasmids were transformed into DH5α competent bacteria (Invitrogen, Carlsbad, California, USA) following the supplier’s instructions. The recombinant plasmids and the inserted sequences were confirmed by Sanger sequencing (Eurofins Genomics, Ebersberg, Germany) with the universal vector primers M13F_uni (-21) (5'-TGT AAA ACG ACG GCC AGT-3') and M13R_rev (-29) (5'-CAG GAA ACA GCT ATG ACC-3'). Recombinant plasmids were amplified, and the quantity and quality were determined using a Synergy™ H1 microplate reader (BioTek, Winooski, Vermont, USA). The DNA copy number was estimated using NEBioCalculator (https://nebiocalculator.neb.com/#!/dsdnaamt), then 7 concentrations with known DNA copy numbers/µl were prepared by serial dilutions and used to estimate the virus copy number in infected and non-infected mosquito samples. Sterile, nuclease-free water was used as a no template control (NTC), then tested in triplicates.
One-step real-time RT-qPCR
Mosquito-borne virus-specific primers and TaqMan probes previously reported to detect each specific virus were synthesized by Eurofins Genomics with 5-FAM, HEX as the reporter dye for the probe. The details of the primers and probes sequences and their characteristics are shown in Table 2. The real-time RT-qPCR assay was performed using a CFX96 Real-Time System cycler (Bio-Rad, Hercules, California, USA) and the Quantitect Probe RT-qPCR kit (Qiagen). Reactions were performed in a 20 µl volume mixture containing 1 µl of RNA template, 10 µl of 2× QuantiTect Probe RT-qPCR Master Mix, 8 µM of forward primer, 8 µM of reverse primer, 2.5 µM of probe, 0.25 µl of QuantiTect RT Mix and 5.95 µl RNase-free water. The amplification of the cDNA and the quantification of the viral copy number was done in one step using the following protocol: a single cycle of reverse transcript for 15 min at 50 °C, 15 min at 95 °C and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The real-time data were analysed using the CFX manager software provided by Bio-Rad. Negative and positive controls were included in all PCR reactions performed. A sample was determined empirically to be positive if the Cq value was 36, based on background cross-reactivity of the primers and probes in non-template control reactions. Positive results were determined according to the amplification cycle at which the relative fluorescence unit (RFU) was detected below the quantification cycle (Cq). Baseline thresholds for the two fluorophores were determined with the CFX manager software in a series of reactions using the virus standard dilutions and then set for subsequent runs as auto calculated [27].
{kind=link}