To fill the unmet need for effective COVID-10 treatments, we leveraged the Library of Integrated Network-based Cellular Signatures (LINCS) - the largest body of drug-induced transcriptional perturbations profiles available to date, the healthy lung transcriptome from the Genotype-Tissue Expression (GTEx) project and the recently published SARS-CoV-2 induced transcriptomic profiles from in vivo and in vitro settings (All settings are described in the method section and illustrated in table 1).
Hallmarks of SARS-CoV-2 infection across in vivo and in vitro settings
In order to address pharmacological gaps in SARS-CoV-2 research, it was of paramount importance to understand the molecular mechanisms underlying the SARS-CoV-2 infection. To this end, we have collected public SARS-CoV-2 transcriptome-wide datasets and normal lung transcriptomics from GTEx. Subsequently, we conducted a differential expression analysis to compare SARS-CoV-2 conditions with their respective control groups. Next, all genes from the different comparisons were merged by the shrunken log2 fold change values (no-cutoff) to yield a full matrix corresponding to 7433 genes x 7 contrasts. These are referred to as SARS2_BALF_WUHAN1-2, SARS2_LUNG, SARS2_NHBE, SARS2_A549_ACE2,SARS2_Calu3 and SARS2_A549_ACE2_RUXO (ruxolitinib-treated cells).
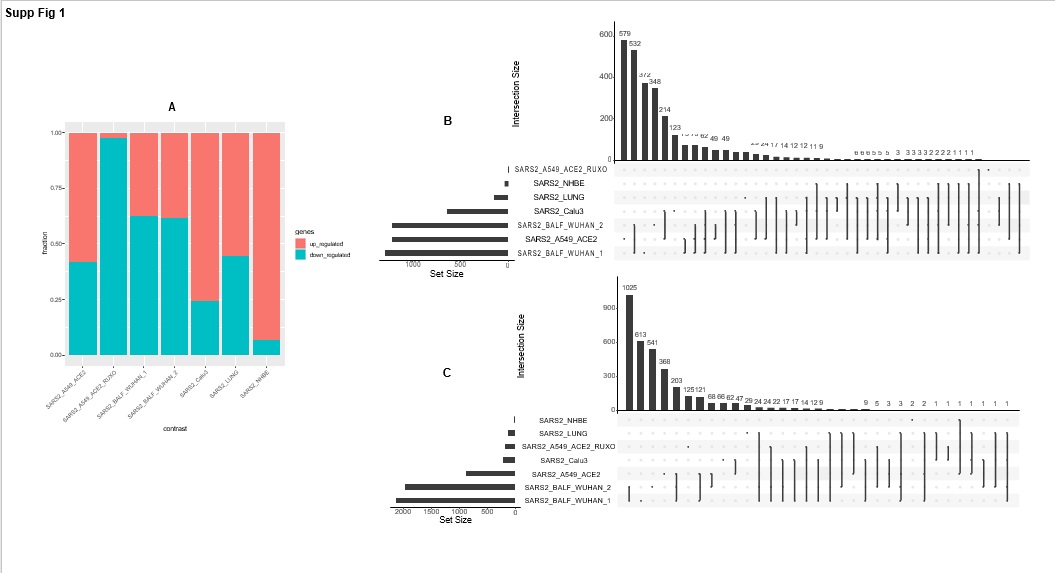
For each of these seven contrasts, the fraction of significant up- and down-regulated genes (|Log2FoldChange| > 1 and p-adjusted-value < 0.05) and the corresponding overlap between contrasts is given in Supplementary Figure 1. As for the transcriptomic data from the Genotype-Tissue Expression (GTEx) project (N=374 samples), gene-wise Pearson correlation of the ACE2 gene and all other expressed genes is referred to as ACE2_GTEX, (Pearson correlation ∈ [-0.47, 0.59]), out of which 7427 genes were shared with the seven SARS-CoV-2 contrasts (Supplementary Table S2) and genes were ranked accordingly.
The ranked lists of genes across all SARS2 contrasts as well as genes ranked by their correlation coefficient from ACE2_GTEX were investigated at the pathway level using gene set enrichment analysis (GSEA). We assessed enrichment against a collection of hallmark genesets and a curated set of genes associated with SARS_CoV infection from the literature (See methods section and Table S3). To assess the similarity between settings, we first computed all pairwise correlations of pathway enrichment scores from all GSEA analysis (Figure 2A). Interestingly, both SARS2_A549_ACE2_RUXO and ACE2_GTEX were strongly concordant (rho= 0.63, p< 0.01). This suggests that ruxolitinib treatment in ACE2- expressing A549 cells and ACE2 baseline expression in the lungs engage a similar set of molecular pathways. Indeed, similarly to ACE2_GTEX, Ruxolitinib-induced pathways were highly anti-correlated with those from all the SARS2 settings (Fig 2A). Although SARS2_LUNG was correlated with BALF settings, this correlation was not significant (p-value > 0.05, Fig 2A) suggesting a functional diversity in response to SARS-CoV-2.
We further investigated the enriched hallmarks/pathways in both the normal lung and SARS-CoV-2 infected in vivo and in vitro settings. By keeping all gene sets with a normalized enrichment score |NES| greater or equal to 1 and a p-adjusted-value threshold of 5%, our analysis identified a highly conserved hallmark process, the TNF Alpha (Tumor necrosis factor-alpha) signaling via NF-κB. This hallmark was significantly upregulated by the SARS-CoV-2 infection but downregulated in both ruxolitinib-treated and ACE2_GTEX groups (Figure 2B). Moreover, to investigate the pathogenic cellular response of SARS-CoV-2, we proceeded as follow: (1) We extracted the union of all leading-edge genes from the conserved hallmark and (2) We extracted leading-edge genes belonging to the following enriched hallmarks: INTERFERON GAMMA RESPONSE, INTERFERON ALPHA RESPONSE, INFLAMMATORY RESPONSE, IL6-JAK-STAT3 signaling and the curated set of SARS-CoV associated genes from the literature (geneshot_SARS_CoV). It is noteworthy to be mentioned that enriched hallmarks from (2) were negatively scoring genesets in ACE2_GTEX and positively enriched in all SARS2 settings but the WUHAN_BALF samples (Figure 2B). Across all settings, the resulting genes were assigned either the shrunken log2 fold change from their corresponding DEG analysis or the correlation value for ACE2_GTEX (Figure 2C). From this collection of relevant genes of innate immunity, we identified a set of conserved upregulated genes in SARS2 settings (group1, Figure 2C). These same upregulated genes were downregulated in ruxolitinib-treated cells and highly anti-correlated with reference to ACE2_GTEX. Most of these genes (e.g. CXCL10, CXCL11, OAS1, TNFSF10, MX1, DDX58, ISG15, IFIT2, IFIT3, EIF2AK2) are induced in
response to interferons. Notably, the induction of these genes was much more pronounced in SARS2_LUNG, SARS2_Calu3, and SARS2_A549_ACE2 (Figure 2C). For instance, the pro-inflammatory chemokine CXCL10/IP-10 had a 32 fold-increase in SARS2_LUNG compared with a 3 fold increase in the WUHAN_BALF samples, and it was almost absent in NHBE infected cells. Chemokine's expression in the postmortem lung sample (SARS2_LUNG) was similar to what is being observed in A549_ACE2 and Calu3 cells infected with high loads of virus (MOI:2). Although MOI of 2 was also used for NHBE cells, RNA-seq profiling 24 hours post-infection showed that these cells efficiently cleared the virus (no change observed for CXCL10-11). In contrast to groups 1 and 2 that were predominantly represented by interferon-induced chemokines and genes regulated by NF-κB in response to TNF alpha, group 3 seems to represent a set of genes belonging to the IL6-JAK-STAT3 pathway (CSF1, IFNGR2, IL4R, IFNGR1, HMOX1, TYK2, TGFB1, TNFRSF1A) and inflammatory responses (PTGER4, IRS2, MAP2K3, AXL, C5AR1, PTAFR); these genes were antagonized by SARS-CoV-2 specifically in BALF samples and not the cell lines. Lastly, group 4 showed a similar pattern to group 1 and its gene members are also related to the Interferon alpha/beta/gamma, and complement signaling (e.g., TRIM25, UBE2L6, STAT1, JAK2, IL1B, IL2RG, GBP2, PLAUR). For instance, STAT1, a known master regulator of interferon genes, has more than a 5 fold-induction in SARS2_LUNG, A549, and Calu3 but its expression was not tangible in the other in vivo settings or SARS2_NHBE. In summary, expression dynamics from the deceased patients are highly similar to those from SARS2_A549_ACE2 and SARS2_Calu3 but dissimilar to WUHAN_BALF and NHBE cells. The former elicits marked pro-inflammatory events which could explain the development of severe cases of COVID-19 symptoms and lung injury as seen in the postmortem samples.
Identification of new COVID-19 repurposing opportunities through mining L1000 connectivity map
In the previous section, we highlighted molecular vulnerabilities providing a rationale for targeting hyperinflammatory response in COVID-19. Although ruxolitinib provided a proof of concept that abrogating inflammation is an appealing therapeutic strategy, we conducted a full data-driven computational approach to identify potential drug candidates that, in addition to ruxolitinib, could perturb the pathological mechanism induced by the new SARS-CoV. To this end, we have interrogated the L1000, the largest repository of chemical-induced gene perturbation profiles. The curation and pre-filtering of the L1000 dataset used in this study are described in the methods section. Briefly, we have assembled expression perturbations of 978 landmark genes across 4487 chemical compounds from LINCS L1000. 727 out of 978 genes (~75%) were shared with our SARS2 and ACE2_GTEX settings. To identify potential drug repurposing candidates, we used the cosine similarity distance between vectors of gene expression signatures. For each of the drug-induced expression profiles, a cosine score was calculated against all SARS2 and ACE2_GTEX settings (shinyApp table provided upon request). A positive cosine score corresponds to similar expression patterns (useful to identify drugs inducing similar modes of action/molecular mechanisms) while a negative score would suggest an opposite expression pattern (likely beneficial in the context of a disease, specifically COVID-19).
We first validated our approach on ruxolitinib signature in SARS-CoV2 infected cells (SARS2_A549_ACE2_RUXO). Our pipeline could accurately and unbiasedly identify Ruxolitinib from L1000 as the second-best matching hit out of 4,487 chemical compounds (cosine score= 0.18, p-value from 10,000 random permutations = 2e-04).
To further ascertain that our approach can identify not only ruxolitinib but its drug target class, we performed a drug set enrichment analysis. In this setting, a drug set is a target gene/protein that is associated with at least 3 drugs. We first ranked drugs by the cosine value and ran the enrichment analysis against 295 target genes (see method section for drug-target preparation and database). With respect to SARS2_A549_ACE2_RUXO, we could confirm that indeed JAK2 inhibitors were significantly overrepresented at the top of the ranked list of cosine scores (ES= 0.825, adjusted p-value=0.08) (Figure 3C).
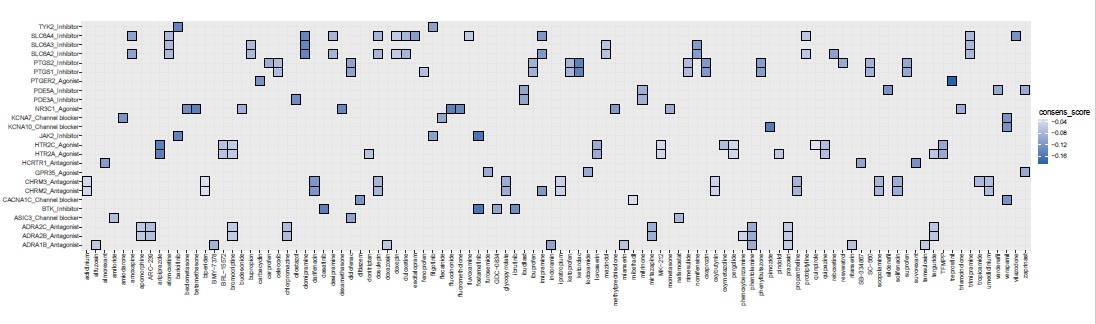
Next, we investigated drug-target classes that display a negative enrichment score (their drug members likely reverse the phenotype) against a computed consensus cosine score which is the median value of the cosine spanning 4,487 drugs across all SARS2 settings. We postulate that the consensus score would emphasize drug classes that target generic processes induced by SARS-CoV-2 across diverse settings and infectivity phases/severity levels. Furthermore, drug-target enrichment analyses were conducted separately for each of the SARS2 settings (excluding RUXO group). (Figure 3A).
Importantly, when considering an enrichment drug score (ES) less or equal to -0.5 and adjusted-p-value < 0.25 (Figure 3A), our consensus signature exhibited the strongest enrichment for JAK2 inhibitors (filgotinib, baricitinib, fostamatinib), Bruton kinase-BTK inhibitors (GDC-0834, ibrutinib, dasatinib; Figure 3D), CACNA1C blockers (mibefradil, diltiazem,verapamil), HCRTR1 (orexin) antagonists (SB-334867/almorexant/suvorexant), PTGER2 agonists (carbacyclin, treprostinil),KCNA7 Channel blockers (amiodarone, flecainide), ASIC3 Channel blocker (amiloride, nafamostat, diclofenac), PDE5A inhibitors (milrinone, zaprinast, vardenafil, ibudilast, sildenafil) and ADRA1B antagonists (prazosin, tamsulosin, alfuzosin, doxazosin, mianserin, ritanserin, phentolamine, indoramin). (Figure 3A). When leveraging the drug enrichment analysis for each of SARS2 settings, we observed that SARS2_LUNG exhibited very few enriched drug classes. Among these enriched classes, BRD4 inhibitors (BI-2536, colchicine) and NR1I2 agonists (hyperforin, mifepristone, nifedipine, clotrimazole, paclitaxel). Interestingly, corticosteroid agonists (methylprednisolone, triamcinolone, budesonide, mometasone, beclomethasone, dexamethasone, fluocinonide, betamethasone, fluorometholone) were enriched in both WUHAN_BALF, consensus, and NHBE settings.
Given the important role of ACE2 in both SARS-CoV-2 infectivity and normal lung physiology, we next sought to identify drugs that could counteract the SARS-CoV-2 invasion by restoring the baseline co-expression network of ACE2 in the healthy lung.
In this particular analysis and similar to SARS2_A549_ACE2_RUXO, a positive enrichment score is the preferred outcome (Figure 3B). A drug-target class with an enrichment score equal or greater than 0.5 is considered a putative candidate to maintain a normal ACE2 expression whereas a drug-target with a negative enrichment score is likely to be beneficial for the virus. The search for such modulators identified drug-targets highly enriched for inhibitors of cereblon protein (lenalidomide, pomalidomide, thalidomide; ES=0.93, adjusted-p value=0.05) and Bruton Kinase BTK (fostamatinib, ibrutinib, dasatinib, GDC-0834; ES=0.86, adjusted-p value=0.023). Strikingly, Angiotensin-converting enzyme inhibitors (enalapril, fosinopril, captopril, perindopril) also showed positive connectivity with ACE2_GTEX (ES=0.61, adjusted-p value=0.22). This could be explained that the Angiotensin-converting enzyme (ACE) expression from GTEx was highly anti-correlated with ACE2 (rho = -0.47). Last, and similar to Bruton kinase inhibitors, we identified drug-target classes such as NR3C1 agonists (corticosteroids), PDE5A inhibitors (milrinone, zaprinast, vardenafil, ibudilast,sildenafil), ADRA2B antagonists (phenoxybenzamine, prazosin, bromocriptine, apomorphine, terguride, ARC-239, chlorpromazine, mirtazapine, phentolamine), HTR2A agonists (MK-212, pergolide, pindolol, terguride, donitriptan, quipazine, lorcaserin, aripiprazole), and KCNA10 channel blockers (verapamil, pimozide). These classes might have a dual effect by modulating the inflammatory response, protecting the lung, and restoring ACE2-induced downregulation by the virus (Figure 3E). We are aware that only a limited fraction of the drugs is assigned to a target class (1030 out of 4487 drugs; 23%). As such, all results from our analyses will be made publicly available to accelerate the search for COVID-19 treatments.
Pathways/molecular hallmarks associated with potential modulators of COVID-19 To understand the mechanism by which the candidate drug candidates block the molecular events underlying COVID-19, we focused on significant drug-target lists associated with the previously described consensus score. This involved 25 drug-target families and an ensemble of 106 drugs (Supplementary Figure S2). Given the small number of genes from the L1000 dataset (727 out of the 978 landmark genes are shared with our data), we opted out to choose gene set enrichment analysis but instead, we conducted a row-wise Welch's t-test to identify the landmark genes that show significant changes between the 106 drugs and the SARS2 groups. This analysis yielded a set of 124 genes that displayed extreme values from the two-tailed t-test (False discovery rate < 5%, 73 genes with positive t-stat values genes, 51 genes with negative t-stat values)(Figure 4A).
When performing a hypergeometric test, separately for the negatively and positively modulated genes considering the set of significant genes, we showed that several down-regulated processes were enriched for Cytokine Signaling, Jak-STAT signaling, Complement activation, TNF-alpha effects on cytokine activity, lung fibrosis and inflammatory response. Gene members include transcripts such as RALA, CD40, FRS2, CRKL, SOCS2, UBE2L6, STX4, SMAD3, CCL2, IFNAR1, SPRED2, PIK3CA, IKBKB(Figure 4B). In contrast, up-regulated processes were enriched for metabolic processes, mainly glucose metabolism, cell cycle, and mRNA processing. Such transcripts include CTD, LBR, PGM1, NUP85, PFKL, PLCB3, PSMD10, SLC5A6, CAT, NUP133, HK1, HSD17B10, ETFB, NUP62, ADH5, NUP88, NNT, RPN1, GNAI2, VAPB, SACM1L (Figure 4C). Finally, although L1000 contains a limited set of genes and lots of transcriptome-wide information is missing, we could confirm that the identified drugs interfere with the hallmarks of the SARS-CoV-2 infection described earlier. More importantly, most of these drugs are FDA approved and patients would benefit from them before reaching severe courses of COVID-19.
{kind=link}
{kind=link}