COVID-19 has become a threat to individual health, national and global health-care systems, and economic and social systems.
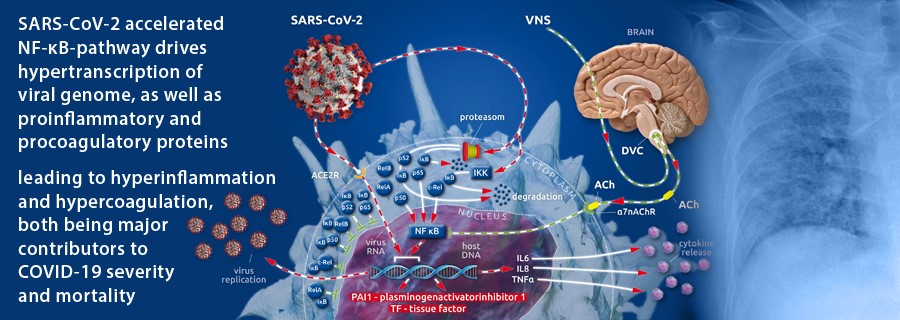
Recently, nuclear factor-κB (NF-κB) (see box I(1)) was shown to play a pivotal role in the pathogenesis of the pulmonary damage and systemic inflammation common to COVID-19(1). A transcriptional factor of pro-inflammatory cytokines (IL6, IL8, TNFα), NF-κB is excessively overexpressed by viral hijacking of its transcription-promoting pathway(1,2).
After cellular invasion via ACE2 receptors(3), SARS-CoV-2 amplifies the action of the cellular IκB kinase complex (IKK)(2), which catalyzes the proteasome-based degradation of cytosolic inhibitors (IκBs)(4) of NF-κB subunits (p65, p50 or p52; c-Rel, RelA or RelB) leading, ultimately, to unrestricted subunit passage through the nuclear membrane and the excessive formation of mature NF-κB(2)(see box II(1)). This, in turn, promotes the uncontrolled downstream transcription of NF-κB-dependent proteins (i.e., SARS-CoV-2 and proinflammatory cytokines)(1) (Figure 1).
The physiological restraint mechanism of cellular NF-κB action is the vagally driven cholinergic anti-inflammatory pathway (CAP)(5–8). This pathway limits NF-κB activity via membrane-bound nicotinic acetylcholine receptors (α7nAChR), which, after coupling to vagally secreted acetylcholine (ACh), initiate blocking of NF-κB-mediated transcription(5–8). Thus, the balance between pro- and anti-inflammatory effects creates a health-preserving immune response to external assault(5–8). In severely ill patients, vagal signaling is critically compromised, leading to inappropriate inflammatory responses(1). This autonomic shift is also seen in COVID-19(1,9). Moreover, SARS-CoV-2 is suspected to bind not only to ACE2 receptors but also to nicotinic acetylcholine receptors (nAChRs)(10). Because α7nAChRs are a pivotal part of the anti-inflammatory reflex arc (involving CAP), virus-related α7nAChR impairment potentiates the NF-κB pathway acceleration evoked by vagal dysfunction and viral hijacking (Figure 1).
In almost all COVID-19 cases, we see elevated coagulation parameters, such as increased d-dimer(11–14) and prolonged prothrombin time (PT)(12–15), as well as prolonged activated partial thromboplastin time (aPTT)(12,13,15). Hence, disseminated intravascular clot formation can be observed(12,13,16–18) in critically ill COVID-19 patients, whereas incidences of significant hemorrhagic complications are rarely reported(16). Several specific aspects of COVID-19-associated coagulopathy (CAC)(15,17) differ from what we know about disseminated intravascular coagulation (DIC)(16). The most important differences between CAC and DIC are the elevated fibrinogen(12) serum levels, the activated complement system(19,20), and increased concentrations of antiphospholipid antibodies(12) during CAC, compared with the lower fibrinogen concentrations, reduced complement system activity, and lower antiphospholipid antibody serum levels seen in DIC pathology(15). Both CAC and DIC share elevated d-dimer values, which reflect fibrinolytic activation in response to venous thromboembolism (VTE)(21). These specific coagulopathic changes in CAC and the high incidence of thromboembolic complications in COVID-19 have led to the proposed use of high d-dimer values as illness severity factors(22) and indicators of a poor prognosis in COVID-19 cases(23). The reasons for the differing specificity of CAC compared with DIC have remained unclear(13,15,16,24,25) until recently.
In our recently published article, we described the importance of the accelerated NF-κB pathway in the pathology of SARS-CoV-2-evoked COVID-19(1). Here, we focus on the fact that procoagulant tissue factor (TF)(24,26–28) and antifibrinolytic plasminogen activator inhibitor 1 (PAI 1)(26,28,29) are transcribed in an NF-κB-dependent manner(26,28). The promotional role of NF-κB in the transcription of two factors (that potently move the hemostatic balance towards clot formation and impaired fibrinolytic reaction) and the viral-induced, specifically accelerated NF-κB pathway are the first reasonable explanations for the specific coagulopathic pattern seen in COVID-19 (Figure 1).
Ventilation-derived oxygen and stretch collaborate with SARS-CoV-2 to activate the NF-κB pathway:
In their review, Jacobson and Birukov (2009) pointed out that, in critically ill patients, mechanical ventilation with hyperoxic gas concentrations is often necessary to secure adequate oxygen delivery via an injured lung to the vital organs(30). Moreover, in most cases, the consolidated lung requires high peak pressure values, even with the use of low tidal volumes (LTVs) due to severely decreased pulmonary compliance(30). High peak pressures with concomitant pulmonary stretch stress together with hyperoxic gas mixtures also result in the activation of nf-κB(30), which is known to promote the transcription of PAI 1 and TF(28,31). Impaired alveolar membrane function combined with severe decreased pulmonal compliance are found in the COVID-19-affected lung(32). Thus, viral acceleration of the NF-κB pathway(1,2), along with the almost unavoidable mechanical and hyperoxic stresses to pulmonary tissue, leads to enhanced NF-κB action. Through increased fibrin clot formation and severely impaired fibrinolytic potential, the concomitant extravascular activation of antifibrinolytic PAI 1 and procoagulant TF leads to micro- and macrovascular thrombosis(30). These, together with the NF-κB-driven enhancement of virus replication(1,2) and further release of proinflammatory cytokines(1,2,33), explain why this avalanche-like process is observed in critically ill COVID-19 cases.
Thrombotic assault in lung and liver:
In their recently published autoptic investigation of COVID-19 fatalities, Lax et al. (2020) described widespread occlusions within the pulmonal arterial vasculature and hepatic vessels, whereas the injury patterns in other parenchymatous organs were predominantly unrelated to malperfusion(17). They stated that these pulmonary arterial occlusions were of thrombotic instead of embolic origin(17). As in the case reported below, all cases presented by Lax et al. received anticoagulant or antiaggregant therapy or a combination of both(17). While in the report by Lax et al., patients received only prophylactic anticoagulant or antiaggregant therapy, every COVID-19 patient in our hospital is medicated with therapeutic anticoagulation from day one of admittance, including the case reported below. Pulmonary thrombotic damage was demonstrable in all samples (n = 11) reported by Lax et al. and ranged from focal to extensive(17). Although pulmonary infarction due to thrombosis occurred in only three of the investigated cases, the authors described the amount of thrombotic material in the pulmonary flow path as gross(17). Additionally, several authors have reported the association between COVID-19 and diffuse and gross clot formation in the branches of the pulmonary artery(18,34–38). Although pulmonary coagulopathy with fibrin clot formation in the lungs due to NF-κB-activated membrane-bound TF and PAI 1(28) is typical in infectious conditions, the specific SARS-CoV-2-related avalanche-like acceleration of the NF-κB pathway seems to influence the amount of enhanced fibrin clotting and fibrinolysis impairment.
In addition to the hypoxic damage caused by thrombosis, investigators have described a plethora of pathological changes, including edema, hyaline membrane, proliferation of pneumocytes and fibroblasts, macrophages, lymphocytes, plasma cells, neutrophils, and collagen, with a heterogeneous distribution(17,18,39,40), which are summarized as diffuse alveolar damage (DAD)(17). The amount of thrombus consolidation is correlated with the duration of the disease(17). Thrombotic clots were detectable in small to mid-sized pulmonary arterial vessels, whose walls were infiltrated by neutrophils, and the adjacent lung tissue showed hemorrhaging and infarction(17). Even in one patient with grossly recognizable pulmonary infarct demarcation, Lax et al. (2020) considered the occlusion to be of thrombotic rather than embolic origin due to the completely thrombotic clot-filled lumen of the affected pulmonary arterial vessels(17). The absence of clot fixation to the arterial wall, as well as the fact that histological consolidation of some of the clots had not yet occurred, led the authors to estimate that these clots formed just a few hours(17) before death. The investigation by Lax et al. (2020) of hepatic tissue in COVID-19 fatalities showed there was massive hepatocyte necrosis in 73% of the cases from chronic congestion of single cells (predominantly pericentral) or a focal or panlobular pattern(17). Finally, in one case, central hepatic vein thrombosis was described(17). Moreover, other histological hepatic changes, such as steatosis (70%), cholestasis with canalicular bile plugs (62%), and portal changes (lymphocytic infiltrate, mild nuclear pleomorphism of cholangiocytes, and ductular proliferation with portal or incomplete septal fibrosis) were seen in nine (82%) of their subjects(17).
{kind=link}