The long noncoding RNAs have been uncovered to exert an pivotal influence on cell proliferation and apoptosis of AML, the mechanisms of which include altering methylation status of gene promoters[5, 6], recruiting epigenetic complex on gene promoters[26], reshaping chromatin [27, 28], sponging miRNAs to regulate gene expression[29-32], etc. HOTAIR is one of the most studied lncRNAs in AML, which is upregulated in de novo AML patients and predicts an adverse prognosis[33]. HOTAIR locates in HOXC gene cluster on chromosome 12 and exerts biological effect through modulating HOXA family genes. Intriguingly, through analysis of TCGA expression data, we found LINC00649 was also correlated with most of HOXA family genes, which encoded crucial transcription factors in normal hematopoiesis, pathogenesis of AML and resistance to chemotherapy[34-36].In comparison with healthy controls, AML patients have aberrantly lower LINC00649 expression in our results. Meanwhile, for most of cancers other than AML, expression level of LINC00649 in cancer cells is higher than that of corresponding normal tissues. Furthermore, the survival (OS and PFS), of LINC00649-low group, was significantly worse than that of LINC00649-high group. The unusual expression signature and prognostic value of LINC00649 drove us to explore the possible molecular mechanisms and uncover its biological function.
According to catRAPID algorithm, 9 proteins containing 120 sites were identified to be potentially binding to LINC00649. TIAL1, SRSF9, SRSF2, SRSF3 and RBFOX2 were identified to be associated with HOXA gene expression. TIAL1 is the RNA binding protein, which binds to target sites and splice the pre-mRNA alternatively[37, 38]. SRSF9 involves in constitutive mRNA splicing and can modulate the target of alternative splicing[39]. SRSF9 was reported to be involved in the cell proliferation and apoptosis in bladder and cervical cancer[40, 41], and related to prognostic alternative splicing events of renal clear cell carcinoma[42]. SRSF2 and SRSF3 are also splicing factors, which belong to serine/arginine-rich protein family. Functional mutations of SRSF2 drive the cancer genesis of hematopoietic cells[43]. SRSF3 is a multiple cancer related splicing factor, namely glioblastoma[44], colon cancer[45], oral squamous carcinoma[46], etc. Moreover, the expression of SRSF2/3 is significantly decreased in de novo AML patients in comparison with that of healthy controls. RBFOX2 can bind to 5’- UGCAUGU-3’ element of target RNA, exerting alternative splicing. RBFOX2 can modulate erythropoiesis, by promoting alternative selection of exon 16 in protein 4.1R, the product of which is essential for erythrocyte membrane stability[47, 48]. Notably, the expression of RBFOX2 is significantly correlated with all members of HOXA family genes (Supplementary Figure 1), suggesting potential interaction between them. Furthermore, the pancancer-TCGA expression data was download from UCSC database (https://xenabrowser.net/hub/), the correlation of RBFOX2 and HOXA genes was analyzed by Pearson’s method (Supplementary Figure 2-1/2/3). Notably, the significant association of RBFOX2 and HOXA is a common feature among cancers generated from different tissues. The expression dataset of normal tissue was downloaded from GTEx database (https://www.gtexportal.org/home/), similar analysis showed that the correlation is insignificant in normal bone marrow (Supplementary Figure 3-1/2/3), which indicated the relationship was a disease-specific feature for AML instead of normal hematopoiesis. All 5 splicing factors and LINC00649 are potential co-regulators for HOXA genes in AML, which has not been explored before.
Based on the results of GESA, the upregulation of PI3K-Akt-mTOR signaling, IL6-JAK-STAT3 signaling, oxidative phosphorylation was identified in LINC00649-low group. The activation of PI3K-Akt-mTOR signaling were found in 50% of AML patients[49, 50]. The PI3K-Akt signaling controls leukemic blast cells proliferation and clonogenicity[51, 52]. Aberrantly functional receptor tyrosine kinases drive the activation of PI3K-Akt-mTOR pathway, including IGF1/IGF1R[53, 54], activated FLT3[55] and DEK-NUP14 fusion protein[56]. The inhibitors of PI3K-Akt-mTOR axis have shown preliminary anti-leukemia effects against AML both in vivo and in vitro [57-63]. The IL6-JAK-STAT3 pathway plays a crucial role in oncogenesis of diverse cancers[64]. Constitutive phosphorylation of STAT3 by autocrine secretion of IL6 is revealed in AML cells[65]. Activation of STAT3 is also uncovered in pediatric AML samples, and the small-molecule inhibitor of STAT3 can induce apoptosis and inhibit formation of blast colonies in vitro[66]. The maintenance of leukemia stem cells depends on BCL2 mediated oxidative respiration, instead of glycolysis as in normal hematopoietic cells[67]. The metformin, targeting oxidative phosphorylation (OXPHOS), induces apoptosis of human leukemia cells in an AMPK-independent way[68]. Cytarabine resistant leukemia cells are characterized by activated OXPHOS, with the high level of reactive oxygen species. Additionally, the resistance can be reversed by agents inducing low OXPHOS status[69]. The p53 signaling and Hedgehog signaling were found to be suppressed in GSEA. Non-mutational p53 dysfunction was common in AML and implicated in diverse inactivating mechanisms[70]. Dysregulation and activation of PI3K-Akt-mTOR signaling pathway can activate MDM2 and interact with NF-kappaB signaling pathway, leading to dysfunction of p53[71]. The activation of PI3K pathway was revealed in LINC00649-low group, which may cause the suppression of p53 signaling and inferior survival considering the central role of p53 in the complex network of AML-associated signaling pathway.
The KEGG analysis showed that the LINC00649-associated genes were enriched in AGE-RAGE, PI3K-Akt, Ras and VEGFR signaling pathways. The AGE and RAGE signaling has been studied in AML, which indicated AGE activated MAP kinase, PI3K and JAK/STAT pathway, leading to proliferation of primary AML samples and AML cell lines[72]. Activation of Ras signaling can also promote the dysfunction of p53 by similar mechanism of PI3K-Akt signaling[71]. VEGFR is reported to be overexpressed[73] in AML, which is in accordance with our results. The activated VEGFR signaling promoted the proliferation, survival and resistance to chemotherapy of AML blasts[74]. VEGFR targeting therapy has been developing and showing preliminary benefit for AML in vitro[75-77]. While the Reactome analysis demonstrated other enriched pathways, including signaling by ERBB2 and VEGFR2 mediated cell proliferation. Mudritinib, an ERBB2 inhibitor, was reported to eliminate AML cell both in vivo and in vitro [78]VEGFR2 is a ‘hot’ target in AML, and relevant to chemotherapy-sensitivity, pro-survival effect and angiogenesis in bone marrow[79, 80]. VEGFR2-targeting therapy is being developed in preclinical stage[80, 81]. The dysregulation of all above pathways contributed to the difference of survival between LINC00649-high and low groups.
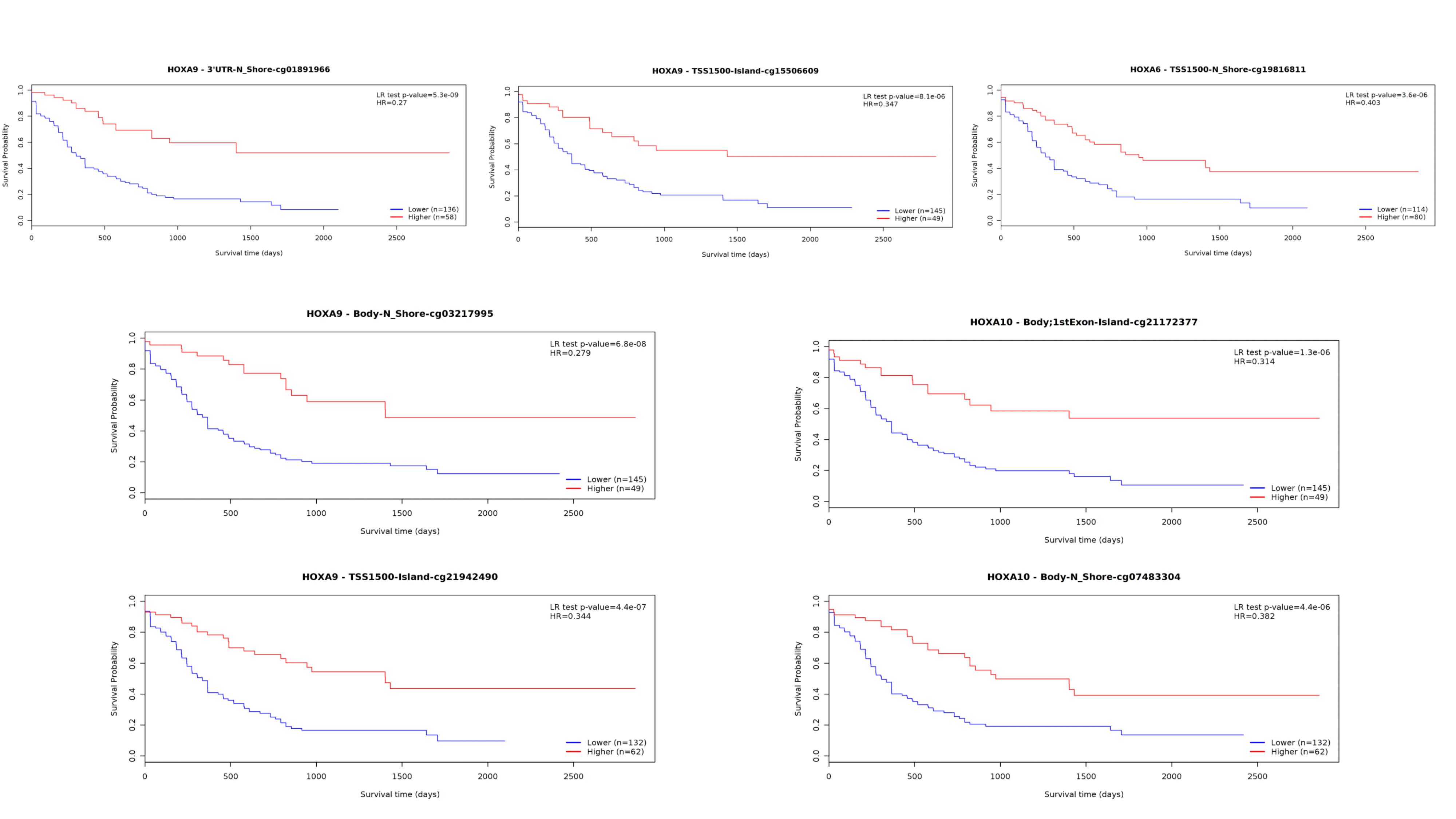
Furthermore, HOXA family genes methylation status was shown to be correlated with LINC00649. The methylation status of seven CpG sites involving with HOXA6/HOXA9/HOXA10 was correlated with expression of LINC00649. Notably, all involved sites were of significance for AML overall survival (Supplementary Figure 4). Considering that lncRNA HOTAIR can modulate the methylation status of HOXA5 by inhibiting DNMT3B[6], our results suggested similar epigenetic mechanism may implicated in the regulation of HOXA genes.
To improve the diagnostic utility, we brought in multi-dimension information to establish a prediction model on AML survival. The traditional prognostic factors (age, gender, ELN2017 risk stratification, etc) and the associated expression data (predicted LINC00649 binding proteins, miRNAs/mRNAs in the ceRNA network), and methylation data (altered methylated CpG sites) were included into the prediction model, by which the OS and PFS data were fitted into using the LASSO regression analysis. A few prediction models, including genetic information of AML patients, have been developed previously, including Clinseq-G[82] (AUC for 3-year OS is 0.730), ELN2017 stratification in the validation cohort [82] (AUC for 3-year OS is 0.65), Li Z et al[82] (AUC for 3-year OS is 0.70), Huang R et al[83] (AUC for 1 year OS is 0.666, AUC for 5 year OS is 0.707), Ha M et al[84] (AUC for 5-year OS is 0.613). The AUC of our prediction models is better than all these models, possibly attributing to the integrated multi-dimension information. On the other hand, the Kaplan-Meier plots supported the risk stratification using our models to divide patients into high-risk and low-risk group, which well recognized the patients with much better prognosis (the median OS of low-risk group has not reached). While due to lack of integrated information in one cohort like TCGA database, which included clinical/RNAseq (protein-coding and noncoding RNA)/miRNAseq/methylation datasets, we can hardly validate this model independently. However, the present work brings clues and insights to further studies, by providing potential biomarker and therapeutic targets.
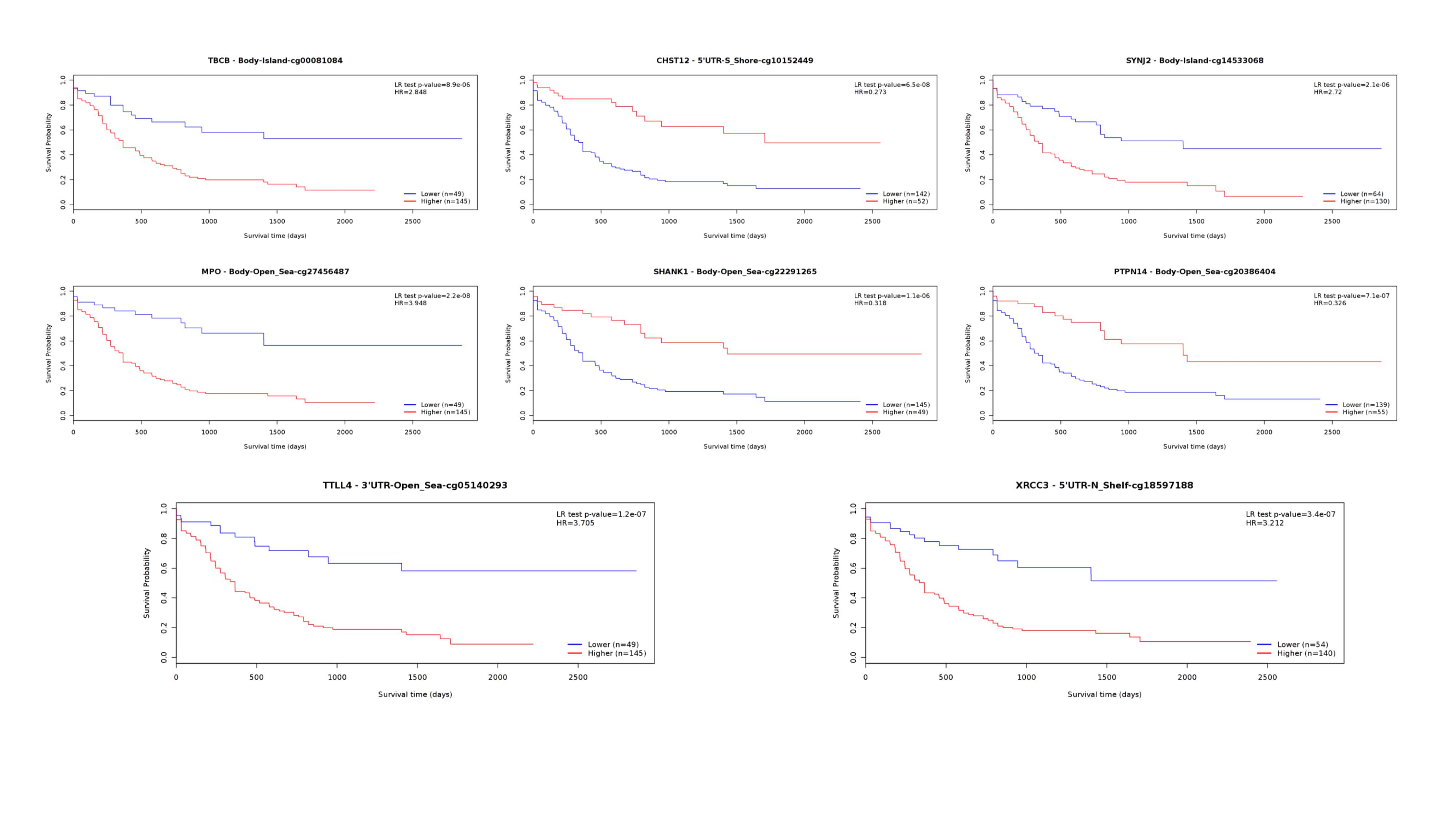
In our OS-prediction model, novel markers were identified (Table 3). EVPL is a component of the cornified envelope of keratinocytes, the genetic variations of which are associated with several solid cancer types[85-89]. While the association of other protein-coding genes or noncoding RNAswith either hematological or solid malignancy has not been investigated. Among the included methylation positions, individual methylation status of cg27456487 (MPO), cg05140293 (TTLL4), cg10152449 (CHST12), cg22291265 (SHANK1), cg18597188 (XRCC3), cg14533068 (SYNJ2), cg00081084 (TBCB) and cg20386404 (PTPN14) were significantly associated with AML survival, according to MethSurv online tools (https://biit.cs.ut.ee/methsurv/) (Supplementary Figure 5). A low expression ratio of MPO has been reported as a deleterious marker for AML, indicating a lower complete remission rate[90] and shorter PFS [91]. In untreated AML patients, hypermethylation status of MPO is detected and correlates with MPO expression, which can be induced by demethylating agents[92]. The alteration of MPO is demonstrated as an indicator for DNA methylation pattern implicating downregulation of DNMT3B[93], our results supported its significance in pathogenesis of AML. Other included methylated sites have not been reported to implicated in AML. In PFS prediction model (Table 3), no genetic variables were described in relation with AML previously. Notably, KIF26A was included in both OS and PFS model, which belongs to kinesin superfamily and is reported as an oncogenic marker for breast cancer[94] and pancreatic ductal carcinoma[95].
{kind=link}
{kind=link}