SA and JA contents changes of M. sieversii responded to the infection of C. mali
The necrotic canker symptom in the wounded twig and leaf infected with C. mali was observed at 5 dpi (Fig. 1a). To measure the changes of phytohormone levels, we implemented the quantitative hormone measurements of JA and SA at 0, 0.5, 1, 2, 3, 6, 24, 48, 120 h infected with the C. mali (Fig. 1b). The production of JA started to increase within 1 h and peaked approximately 10-fold (1262.98 ± 37.76 ng/g FW) at 3 hpi. However, with the increase of the production of SA, the content of JA was reduced accordingly due to antagonistically regulated by the SA from 3 to 6 hpi. Meanwhile, the content of SA was decreased at 3 hpi due to the antagonistic effect of JA. Subsequently, the SA production was increased from 3 to 6 hpi and reached a peak with increased approximately 3-fold (649.10 ± 37.38 ng/g FW) at 48 hpi. From 6 to 120 hpi, the SA and JA presented a consistent pattern such that increased first and then reduced to synergistically respond to the infection. These results imply that the JA-dependent necrotrophic resistance was intensively induced by the invasion of the C. mali. A string of signal transductions and transcriptional regulation processes might be triggered after the infection of C. mali. Additionally, the relative gene expression of key genes of SA and JA synthesis and signaling transduction pathways were detected by qRT-PCR at 0, 0.5, 1, 2, 3, 6, 24, 36 hpi (Fig. 1c). The relative expression level of lipoxygenase 3 (LOX3) and allene oxide cyclase 4 (AOC4) (JA key synthesis genes) were strongly increased after infection, especially the 80-fold higher expression of LOX3 at 1 hpi and about 2000-fold expression of AOC4 from 2 to 3 hpi than 0-hpi control. The gene expression level of coronatine-insensitive protein 1 (COI1) gene, JA signal transduction gene, was slightly reduced after infection. The key SA synthesis genes isochorismate synthase 1 (ICS1) and phenylalanine ammonia-lyases 1 (PAL1) were significantly up-regulated after infection, especially the 300-fold higher expression of PAL1 at 3 hpi. The expression of NPR1, SA key signal transduction gene, was increased from 0.5 to 2 hpi and then decreased after 6 dpi. The pathogenesis-related protein 5 (PR5) and pathogenesis-related protein (PR10) were continuously up-regulated after infection with a 2000-fold higher and 13-fold higher increase than control respectively. These results suggested that JA was induced initially to respond to the infection of the necrotrophic pathogen C. mali.
Sequencing of the M. sieversii transcriptome infected with C. mali using the PacBio platform
To identify and characterize the transcriptomes of M. sieversii twigs inoculated with C. mali during different disease response stages, we employed the PacBio SMRT and Illumina sequence technologies for transcriptome. The PacBio SMRT sequencing yielded all 12,666,867 subreads (25.71G) with an average read length of 2,030 bp, of which 488,689 were full-length non-chimeric reads (FLNC), containing the 5’ primer, 3’ primer and the poly (A) tail (Table 1). The average length of the full-length non-chimeric read was 2,264 bp. We used an isoform-level clustering (ICE) algorithm to achieve accurate 159,249 polished consensuses (Fig. 2a). All these consensuses were corrected using the 1,098,860,548 Illumina clean reads as input data. After error correction and removal of redundant transcript using the LoRDEC, a total of 159,249 non-redundant transcripts were produced, and each represented a unique full-length transcript of average length 2371 bp and N50 of 2596 bp (Table 1). Longer isoforms were identified from Iso-Seq than from the M. domestica reference database (GDDH13 v1.0) and more exons were found in this study (Fig. 2b, c). We compared the 52,538 transcripts with the M. domestica genome gene set, and they were classified into three groups as follows: (i) 11,987 isoforms of known genes mapped to the M. domesitica gene set, (ii) 36,653 novel isoforms of known genes and (iii) 3,898 isoforms of novel genes (Fig. 2d). In this study, the high percentage (69.76%) of new isoforms were identified by PacBio full-length sequencing. It suggested that the high percentage of novel isoforms sequenced by SMRT provided a larger number of novel full-length and high-quality transcripts through the correction of RNA-seq.
Table 1
Statistics of SMRT sequencing data.
| Sample | Mix0_5d |
| Subreads base (G) | 25.71 |
| Subreads number | 12,666,867 |
| Average subreads length (bp) | 2,030 |
| CCS | 633,537 |
| Number of 5'-primer reads | 593,825 |
| Number of 3'-primer reads | 591,975 |
| Number of Poly-A reads | 539,418 |
| Number of flnc reads | 488,689 |
| Average flnc read length (bp) | 2,264 |
| Flnc/CCS percentage (FL%) | 77.14 |
| Polished consensus reads | 159,249 |
| Average consensus reads length (bp) | 2,362 |
| After correct average consensus reads length (bp) | 2,371 |
| N50 | 2,596 |
Alternatively spliced (AS) isoform and long non-coding RNA identification
AS events in different canker disease response stages were analyzed with SUPPA software. We detected 15,607 genes involved AS events (RI, A3, A5, SE, AF, AL) of a total of 20,163 isoforms from the Iso-Seq reads. Most AS events in Iso-Seq were retained intron (RI) with several 4,506 (Fig. 3a). The exon position was 13,767,261 − 13,767,364 in chromosome 11 of the reference genome (Additional file 1). To identify accurately differential APA sites in M. sieversii during canker disease response, 3′ ends of transcripts from Iso-Seq were investigated. There was a total of 23,737 APA sites of 12,552 genes with at least one APA site (Fig. 3b, Fig. 4c, and Additional file 2). We also identified 1,602 fusion transcripts (Additional file 3). Moreover, a total of 1,336 lncRNAs were identified by four computational methods from 1,168 genes of Iso-SEq. The length of the lncRNA varied from 200 to 6384 bp, with the majority (54.87%) having a length ≤ 1000 bp. There were 20 annotated lncRNAs using BLASTN and 1,316 novel lncRNAs with a mean length of 1098.78 bp (Additional file 4). We classified them into 4 groups: 233 sense overlapping (17.44%), 392 sense intronic (29.34%), 295 antisense (22.08%), and 416 lincRNA (31.14%) and mapped them to the chromosomes (Fig. 3c, d, and Additional file 4).
Functional annotations and classifications of DETs
To identify key factors involved in the canker disease response stage, we identified 8,139 DETs from 6,577 differentially expressed genes (DEGs) based on Iso-seq data. The heatmap of DETs showed that numerous biotic response genes were also extensively up and down-regulated in the disease stage (Fig. 5a). Among all these DETs, the most (2,079) and the smallest number of DETs (390) were identified as being differentially expressed in the disease response stage. Using the H-means clustering algorithm, 8,139 DETs were grouped into 6 clusters (H1-H6) (Fig. 5b). Based on the expression changes in disease response stages (1, 2, and 5 dpi), we identified the disease response related to DETs involved in different response stages.
To determine the functions of resistance (R) genes in M. sieversii during the infection of C. mali, the expression patterns of DETs involved in the signaling pathway in plant immunity were analyzed. As well known the microbe-associated molecular patterns (MAMPs) recognized genes Flagellin sensing 2 (FLS2) (MD05G1297700) were significantly increased at 5 dpi. The regulator of chitin-induced immunity the Probable serine/threonine-protein kinase PBL 9 (PBL9) (MD07G1199400) and PBL19 (MD03G1092100 and MD07G1093200) were significantly up-regulated at 5 dpi. Subsequently, the signal transduction related kinase, mitogen-activated protein kinases (MAPKs), mitogen-activated protein kinase kinase kinase 5 (MAPKKK5) (MD15G1035800) was significantly up-regulated at 5 dpi, which was the consistent expression patterns with PBLs (Additional file 5). The expression of dihydroflavonol 4-reductase (DFR) (MD11G1229100) was significantly increased in the defense-related compound flavonoid biosynthesis process from 2 to 5 dpi. The key gene in lignin formation: PAL1 (MD12G1116700, MD01G1106900 and MD07G1172700), caffeic acid 3-O-methyltransferases (COMT1) (MD01G1089800, MD01G1089900, MD07G1161100, MD07G1209500, MD07G1209600, MD07G1300200, MD07G1300500, and MD12G1103500) which were lignin biosynthesis-related genes, were significantly increased after infection. Besides, the peroxidase 51 (PER51) (MD00G1112500), which can generate the reactive oxygen species (ROS) to respond to the pathogen attack, was continually ascended from 1 to 5 dpi (Fig. 5a, and Additional file 5).
To validate the expression pattern of the transcripts in different stages of the canker disease response, we performed qRT-PCR experiments. We selected eight DETs of different aspects of disease response stages with seven DETs showed elevated expression levels and one DET with reduced expression pattern. The qRT-PCR result showed that eight selected DETs have consistent gene expression patterns with RNA-seq data (Fig. 5c). The expression of ethylene-responsive transcription factor 1b (ERF1b) (MD10G1184800) and WRKY33 transcription factors (MD11G1059400) were significantly up-regulated from 2 to 5 dpi. The expressions of the plant resistance (R) genes RPM1-interacting protein 4 (RNI4) (MD05G1172400), pathogenesis-related protein 1b (PR1b) (MD05G1108800), PR5 (MD08G1011900), and glutathione S-transferase 23 (GST23) (MD00G1136300) were significantly up-regulated at 5 dpi compared to 0 dpi. Contrarily, expression levels of the heat shock protein 90 (HSP90) (MD08G1011200) and WRKY70 (MD07G1234700) were significantly down-regulated after infection. This independent qRT-PCR evaluation confirmed the accuracy and reliability of the Illumina sequencing results.
Different regulatory pathways during the response to the infection of the C. mali
Plant defense response to biotic stress involves complex molecular or genetic networks. To further investigate the functions of DETs after the infection of C. mali, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment were implemented (corrected P-value < 0.05).
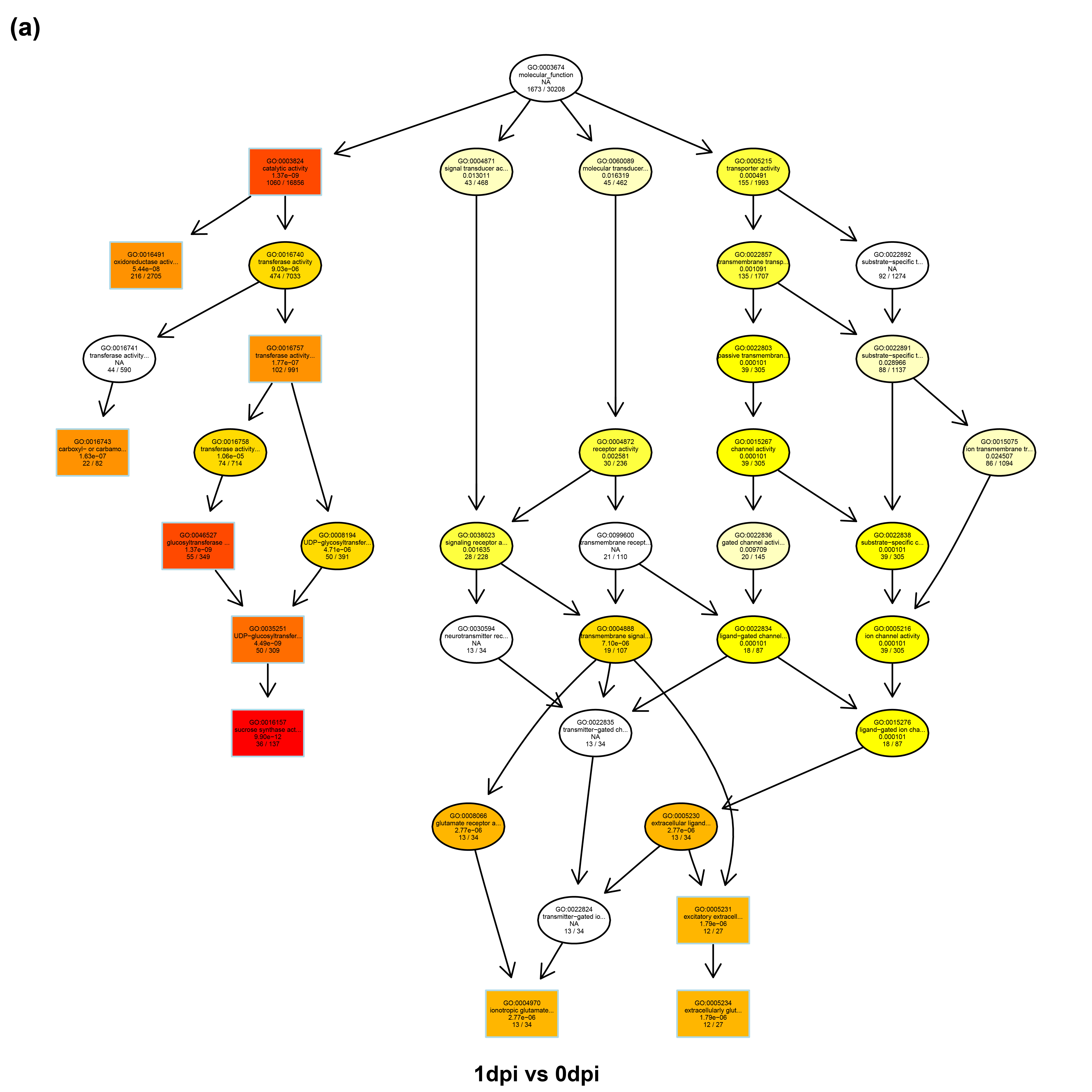
The results showed that the “UDP-glucosyltransferase activity” (GO: 0035251) was significantly differentially enriched with 37 up-regulated transcripts and 13 down-regulated transcripts at 1 dpi (Additional file 6, and 7). The “oxidoreductase activity” (GO: 0016491) was significantly differentially enriched with 201 up-regulated transcripts and 114 down-regulated transcripts at 2 dpi and with 350 up-regulated transcripts and 92 down-regulated transcripts at 5 dpi, including the PER51 (Additional file 6, 8, and 9).
The enriched TOP 20 KEGG pathways of the DETs were showed based on KEGG enrichment, providing transcripts of genes expression overview during the different canker disease response stages. At the early disease response stage (1 to 2 dpi), four pathways were significantly enriched including “plant-pathogen interaction” (ko04626), “starch and sucrose metabolism” (ko00500), “protein processing in endoplasmic reticulum” (ko04141), and “flavonoid biosynthesis” (ko00941) (Fig. 6a, b, Additional file 10). At the late response stage (5 dpi), the pathway “plant hormone signal transduction” (ko04075) was the most significantly enriched with a rich factor 0.156. Moreover, there were the greatest number of genes (86) in this pathway (Additional file 10). Especially, the pathway “phenylpropanoid biosynthesis” (ko00940) was enriched at both early and late response stages (Fig. 6a-c, Additional file 10). It suggested that these pathways played vital and different roles during the response in M. sieversii after the C. mali infection.
To further study the enrichment pathway “plant hormone signal transduction”, the dynamic changes of phytohormone SA, JA, and ET related DETs expression were presented, during the response to the infection of C. mali (Fig. 7). In SA pathway, the necessary key genes Enhanced Disease Susceptibility1 (EDS1) (MD06G1182600, MD14G1188600, and MD14G1188700), Phytoalexin Deficient4 (PAD4) (MD15G1136300) and Senescence-associated carboxylesterase 101 (SAG101) (MD09G1039800, MD17G1039600, MD09G1039700, MD09G1039000, MD09G1038500, and MD09G1038700) in plant systemic acquired resistance (SAR) signal generation and perception were down-regulated from 1 and 5 dpi. However, the marker gene for the SA-mediated signaling pathway PR1b (MD05G1109100 and MD05G1108800) were significantly increased (Fig. 7a). JA synthesis-related key genes, allene oxide synthase 3 (OsAOS3) (MD10G1085800), 12-oxo-phytodienoic acid reductases 3 (OPR3) (MD12G1067300) and Cytochrome P450 94C1 (CYP94C1) (MD11G1171100 and MD14G1019500) were significantly up-regulated from 1 to 5 dpi. The well-known key player in downstream of JA COI1 (MD01G1147000) was significantly increased. The MYC2 (MD06G1034300) core signal transduction mechanism of JA signaling, were significantly up-regulated from 1 to 5 dpi in this pathway. The repressor of JA signaling transduction-related gene Protein TIFY 10A (TIFY10A) (MD02G1096100) was significantly reduced. The key integrator of JA and ET signals in JA/ET dependent defense, ERF1b (MD05G1198700, MD13G1213100, MD10G1184800, MD12G1246000, and MD16G1216900) were significantly increased (Fig. 7b). Meanwhile, ET synthesis-regulated key genes 1-aminocyclopropane-1-carboxylate synthase 1 (ACS1) (MD01G1070400, MD06G1090600, and MD14G1111500) were significantly up-regulated from 1 to 5 dpi. The negative regulator Protein Reversion-to-Ethylene Sensitivity 1 (RTE1) (MD15G1060400) was significantly decreased from 1 to 5 dpi. The receptors of ET, Ethylene response sensor 1 (ERS1) (MD03G1292200), and Ethylene response sensor 1 (ETR2) (MD13G1209700, MD16G1212500, and MD16G1212600) showed highly increased expression levels at 5 dpi. The ET-responsive transcription factor ERF1b (MD05G1198700, MD13G1213100, MD10G1184800, MD12G1246000, and MD16G1216900) were significantly up-regulation from 1 to 5 dpi (Fig. 7c). By combing these dynamic expressions of DETs with the production levels of SA and JA above (Fig. 1c), we determined that the SA and JA antagonistically responded to the infection of C. mali at the early response stage, and synergistically. Nevertheless, the dynamic expression of the key signaling genes in the ET pathway showed a similar expression pattern to that in JA. These results imply that JA and ET might regulate the complex response in M. sieversii to the infection of C. mali.
Transcription factor dynamics during canker disease response stages
TFs and transcription regulators (TRs) play important regulatory roles in the plant response process to the C. mali infection. Totally 3,780 putative TFs, including TFs (2,616) and TRs (1,164) from 88 families were identified and classified with iTAK. The 523 out of these 2,616 TFs were differentially expressed during canker disease response stages. To determine the co-expression and correlation networks of all differentially expressed TFs, weighted correlation network analysis (WGCNA) was conducted (Fig. 8, Additional file 11). Three modules (colored turquoise, brown, and blue) of highly correlated with canker response stage (0, 1, and 5 dpi) were identified (Fig. 8b). Most TFs (140) belong to the turquoise module, in which the peak expression of most TFs was at 0 dpi. The TFs (55) of brown module and TFs (74) of the blue module have decreased and increased expression from 1 dpi to 5 dpi, respectively (Fig. 8c). Family-specific expression was observed in different stages of the canker disease response (Fig. 8d). Correlation of TOP 4 TFs families peaked at 0 dpi, which were bHLH (10), AP2-ERF (7), bZIP(7) and WRKY(7), including bHLH (bHLH4, bLH6, bHLH14, bHLH51, bHLH78, bHLH93, bHLH128, BIM2, PIF3, UNE12), AP2-ERF (ERF9, ERF011, ERF105, ERF106, ERF115, DREB3, RAP2-4), bZIP (bZIP44, CPRF2, GBF4, POSF21, PAN, TGA7, TGA21) and WRKY (WRKY3, WRKY6, WRKY21, WRKY33, WRKY44, WRKY51, WRKY70). Correlation of TOP 5 TFs families peaked at 1 dpi, including Trihelix (5), bZIP (4), and bHLH (4), MYB_related (4), AP2-ERF (3). Among this, they were that Trihelix (ASIL2, AT3G10030.1, AT3G10040.1, GT-2, GTL1), bZIP (CPRF2, GBF4, TGA21, CPRF2), bHHLH (bHLH121, bHLH130, bHLH14, PIF3), MYB_related (MdMYBR1, RVE1, RVE7, TRP6), AP2-ERF (ANT, ERF9, MdERF073). At 5 dpi, WRKY (9), MYB (5), NAC (5) AP2-ERF (4), and HD-ZIP (4) were the TOP 5 highly correlated TFs families, which were WRKY (WRKY6, WRKY7, WRKY19, WRKY33, WRKY40, WRKY45, WRKY51, WRKY61, WRKY75), MYB (MYB4, MYB14, MYB62, MYB108, MYB330), NAC (NAC002, NAC029, NAC045, NAC083, NAC100), AP2-ERF (AIL6, ERF2, ERF114, SlPTI5), and HD-ZIP (ATHB-6, GL2, HAT5, HAT14). Among these, the WRKY family was the most abundant type identified in M. sieversii in response to C. mali infection. These data suggested that TFs in M. sieversii were responsible for pathogen stimulus and interaction with other genes during the host response.
{kind=link}
{kind=link}
{kind=link}