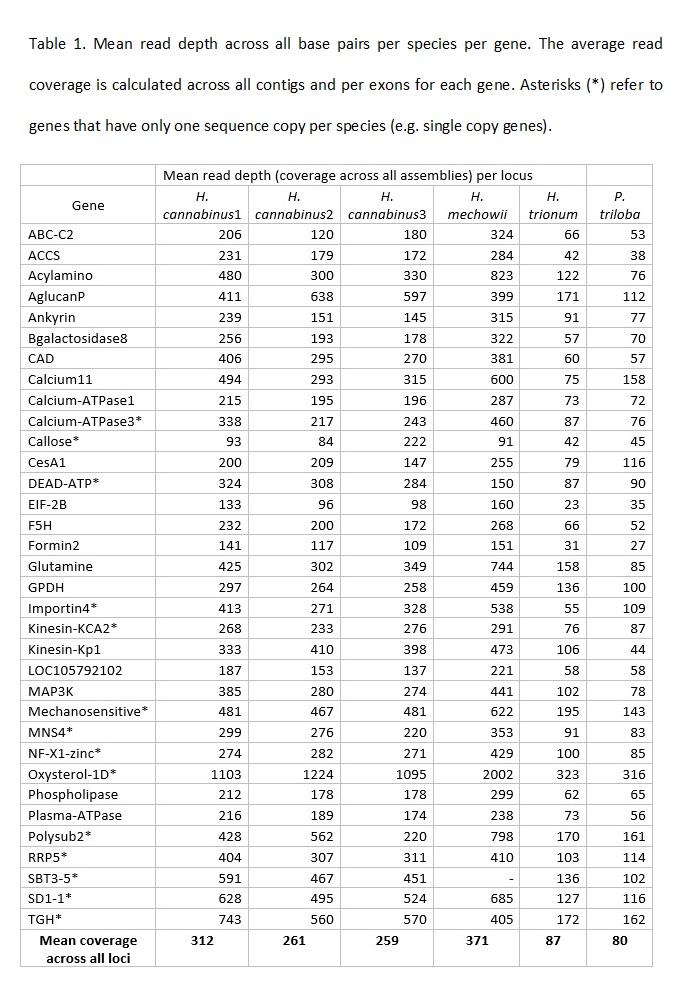
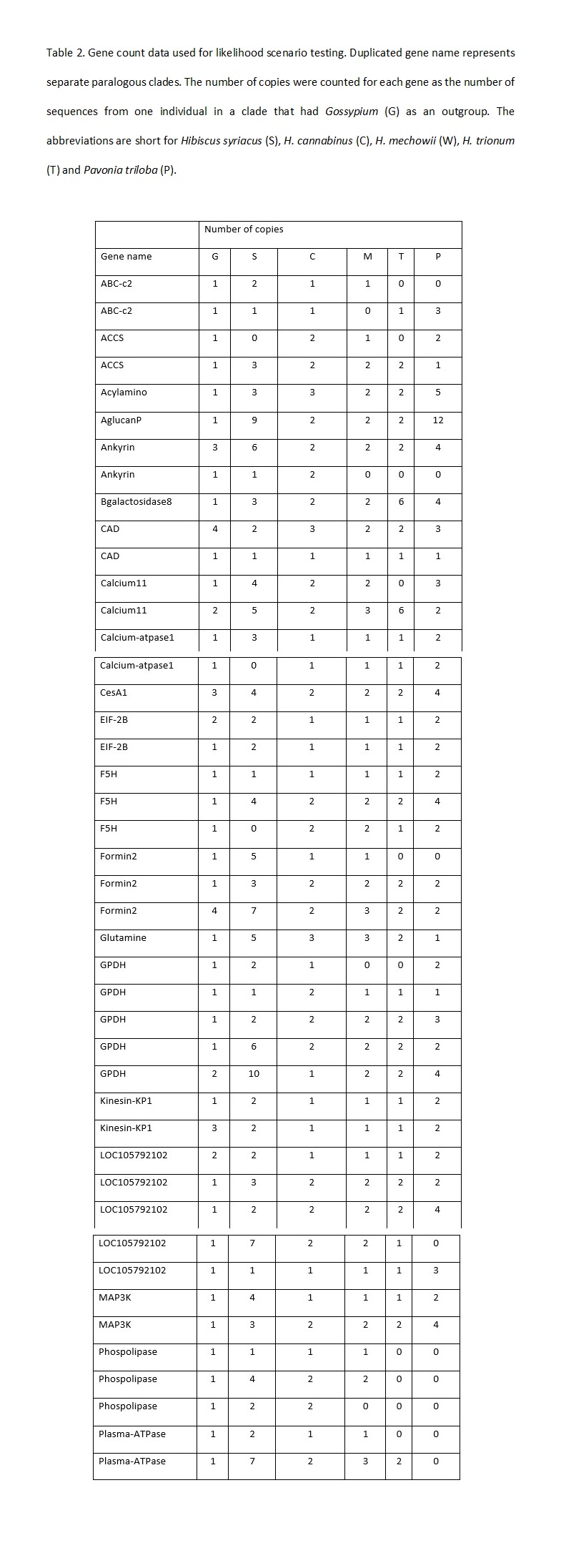
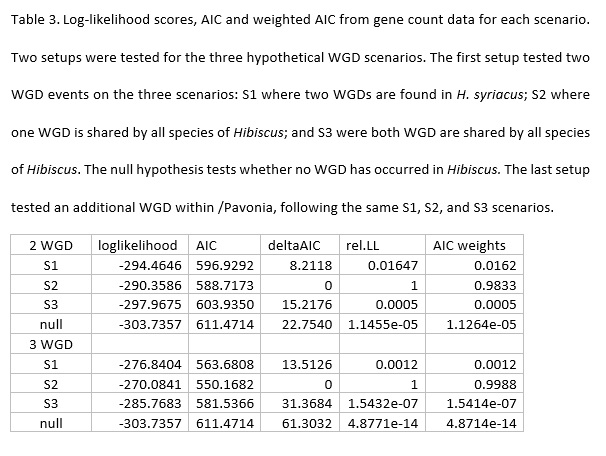
Bates, D. M., 1969 Generic relationships in the Malvaceae, Tribe Malvaea. Gentes Herb. 10: 117-135.
Bates, D. M., and O. J. Blanchard Jr, 1970 Chromosome numbers in the Malvales. II. New or otherwise noteworthy counts relevant to classification in the Malvaceae, tribe Malveae. American Journal of Botany: 927-934.
Baum, D. A., S. DeWitt Smith, A. Yen, W. S. Alverson, R. Nyffeler et al., 2004 Phylogenetic relationships of Malvatheca (Bombacoideae and Malvoideae; Malvaceae sensu lato) as inferred from plastid DNA sequences. American Journal of Botany 91: 1863-1871.
Blanc, G., and K. H. Wolfe, 2004 Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. The Plant Cell 16: 1667-1678.
Borrill, P., N. Adamski and C. Uauy, 2015 Genomics as the key to unlocking the polyploid potential of wheat. New Phytologist 208: 1008-1022.
Bouckaert, R., J. Heled, D. Kühnert, T. Vaughan, C.-H. Wu et al., 2014 BEAST 2: a software platform for Bayesian evolutionary analysis. Journal of PLoS Computational Biology 10: e1003537.
Browning, S. R., and B. L. Browning, 2011 Haplotype phasing: existing methods and new developments. Nature Reviews Genetics 12: 703.
Brummitt, P., and C. E. Powell, 1992 Authors of plant names. Royal Botanic Gardens Kew.
Buggs, R. J., S. Renny‐Byfield, M. Chester, I. E. Jordon‐Thaden, L. F. Viccini et al., 2012 Next‐generation sequencing and genome evolution in allopolyploids. American Journal of Botany 99: 372-382.
Cannon, S. B., L. Sterck, S. Rombauts, S. Sato, F. Cheung et al., 2006 Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proceedings of the National Academy of Sciences USA 103: 14959-14964.
Dasgupta, A., 1981 Cytotaxonomy of Malvaceae II. Cytologia 46: 149-160.
Davie, J. H., 1933 Cytological studies in the Malvaceae and certain related families. Journal of Genetics 28: 33-67.
Doyle, J. J., 1992 Gene trees and species trees: molecular systematics as one-character taxonomy. Systematic Botany 17: 144-163.
Drummond, A. J., S. Y. Ho, M. J. Phillips and A. Rambaut, 2006 Relaxed phylogenetics and dating with confidence. Journal of PLoS Biology 4.
Drummond, A. J., M. A. Suchard, D. Xie and A. Rambaut, 2012 Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 29: 1969-1973.
Fawcett, J. A., S. Maere and Y. Van de Peer, 2009 Plants with double genomes might have had a better chance to survive the Cretaceous–Tertiary extinction event. Proceedings of the National Academy of Sciences USA 106: 5737-5742.
Fernández, A., A. Krapovickas, G. Lavia and G. Seijo, 2003 Cromosomas de Malváceas. Bonplandia: 141-145.
Fryxell, P. A., 1999 Pavonia Cavanilles (Malvaceae). Flora Neotropica Monograph 76.
Fu, L., B. Niu, Z. Zhu, S. Wu and W. Li, 2012 CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28: 3150-3152.
Grant, V., 1981 Plant Speciation. New York: Columbia University Press.
Gregg, W. T., S. H. Ather and W. M. Hahn, 2017 Gene-tree reconciliation with MUL-trees to resolve polyploidy events. Systematic Biology 66: 1007-1018.
Huelsenbeck, J. P., B. Larget and M. E. Alfaro, 2004 Bayesian phylogenetic model selection using reversible jump Markov chain Monte Carlo. Molecular biology and evolution 21: 1123-1133.
Jaillon, O., J.-M. Aury, B. Noel, A. Policriti, C. Clepet et al., 2007 The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463.
Jiao, Y., J. Leebens-Mack, S. Ayyampalayam, J. E. Bowers, M. R. McKain et al., 2012 A genome triplication associated with early diversification of the core eudicots. Genome Biology 13: R3.
Johnson, M. T., E. J. Carpenter, Z. Tian, R. Bruskiewich, J. N. Burris et al., 2012 Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PloS One 7: e50226.
Katoh, K., K. Misawa, K. i. Kuma and T. Miyata, 2002 MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Journal of Nucleic Acids Research 30: 3059-3066.
Katoh, K., and D. M. Standley, 2013 MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772-780.
Kay, K. M., J. B. Whittall and S. A. Hodges, 2006 A survey of nuclear ribosomal internal transcribed spacer substitution rates across angiosperms: an approximate molecular clock with life history effects. BMC Evolutionary Biology 6: 36.
Kearse, M., R. Moir, A. Wilson, S. Stones-Havas, M. Cheung et al., 2012 Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647-1649.
Kim, Y., S. Kim, N. Koo, A. Shin, S. Yeom et al., 2017 Genome analysis of Hibiscus syriacus provides insights of polyploidization and indeterminate flowering in woody plants. DNA Research 24: 71-80.
Koopman, M. M., and D. A. Baum, 2008 Phylogeny and biogeography of tribe Hibisceae (Malvaceae) on Madagascar. Systematic Botany 33: 364-374.
Landis, J. B., Soltis, D. E., Li, Z., Marx, H. E., Barker, M. S., Tank, D. C., and Soltis, P. S.. 2018. Impact of whole‐genome duplication events on diversification rates in angiosperms. American Journal of Botany 105: 348– 363.
Lawrence, W. J. C., 1931 The secondary association of chromosomes. Cytologia 2: 352-384.
Les, D. H., 2017 Aquatic dicotyledons of North America: ecology, life history, and systematics. CRC Press.
Li, H., B. Handsaker, A. Wysoker, T. Fennell, J. Ruan et al., 2009 The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078-2079.
Li, W., and A. Godzik, 2006 Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658-1659.
Marshall, D. C., C. Simon and T. R. Buckley, 2006 Accurate branch length estimation in partitioned Bayesian analyses requires accommodation of among-partition rate variation and attention to branch length priors. Systematic Biology 55: 993-1003.
Matasci, N., L.-H. Hung, Z. Yan, E. J. Carpenter, N. J. Wickett et al., 2014 Data access for the 1,000 Plants (1KP) project. GigaScience 3: 17.
Menzel, M. Y., 1966 The pachytene chromosome complement of Hibiscus cannabinus. Cytologia 31: 36-42.
Menzel, M. Y., and F. D. Wilson, 1969 Genetic relationships in Hibiscus sect. Furcaria. Brittonia 21: 91.
Murray, B. G., L. A. Craven and P. J. De Lange, 2008 New observations on chromosome number variation in Hibiscus trionum s.l. (Malvaceae) and their implications for systematics and conservation. New Zealand Journal of Botany 46: 315-319.
Otto, S. P., and J. Whitton, 2000 Polyploid incidence and evolution. Annual Review of Genetics 34: 401-437.
Paterson, A. H., J. F. Wendel, H. Gundlach, H. Guo, J. Jenkins et al., 2012 Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492: 423.
Pellicer, J., O. Hidalgo, S. Dodsworth and I. J. Leitch, 2018 Genome size diversity and its impact on the evolution of land plants. Genes 9: 88.
Pfeil, B., C. Brubaker, L. Craven and M. Crisp, 2002 Phylogeny of Hibiscus and the tribe Hibisceae (Malvaceae) using chloroplast DNA sequences of ndhF and the rpl16 intron. Systematic Botany: 333-350.
Pfeil, B., C. L. Brubaker, L. A. Craven and M. Crisp, 2004 Paralogy and orthology in the Malvaceae rpb2 gene family: investigation of gene duplication in Hibiscus. Molecular Biology and Evolution 21: 1428-1437.
Pfeil, B., and M. Crisp, 2005 What to do with Hibiscus? A proposed nomenclatural resolution for a large and well known genus of Malvaceae and comments on paraphyly. Australian Systematic Botany 18: 49-60.
Rabier, C.-E., T. Ta and C. Ané, 2013 Detecting and locating whole genome duplications on a phylogeny: a probabilistic approach. Molecular Biology Evolution 31: 750-762.
Ronquist, F., and J. P. Huelsenbeck, 2003 MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572-1574.
Schnable J. C., Springer N. M., Freeling M., 2011 Differentiation of the maize subgenomes
by genome dominance and both ancient and ongoing gene loss. Proceedings of the
National Academy of Sciences USA 108: 4069-4074.
Schranz, M. E., and T. Mitchell-Olds, 2006 Independent ancient polyploidy events in the sister families Brassicaceae and Cleomaceae. The Plant Cell 18: 1152-1165.
Seelanan, T., A. Schnabel and J. F. Wendel, 1997 Congruence and consensus in the cotton tribe (Malvaceae). Systematic Botany 22: 259-290.
Skovsted, A., 1941 Chromosome numbers in the Malvaceae II. Comptes rendus des traveaux du laboratoire Carlberg. Physiologique: 195–242.
Soltis, D. E., V. A. Albert, J. Leebens-Mack, C. D. Bell, A. H. Paterson et al., 2009 Polyploidy and angiosperm diversification. American Journal of Botany 96: 336-348.
Soltis, P. S., D. B. Marchant, Y. Van de Peer and D. E. Soltis, 2015 Polyploidy and genome evolution in plants. Current Opinion in Genetics & Development 35: 119-125.
Stebbins, G., 1947 Types of polyploids: their classification and significance. Advances in Genetics 1: 403-429.
Stebbins Jr, C., 1950 Variation and evolution in plants: progress during the past twenty years. in Essays in Evolution and Genetics in Honor of Theodosius Dobzhansky, edited by M. K. Hecht and W. C. Steere. Springer, Boston, MA.
Tamura, K., and M. Nei, 1993 Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10: 512-526.
Tang, H., X. Wang, J. E. Bowers, R. Ming, M. Alam et al., 2008 Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Research: gr. 080978.080108.
Thomas BC, Pedersen B, Freeling M. 2006 Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Research 16:934-46.
Turner, B. L., and M. G. Mendenhall, 1993 A revision of Malvaviscus (Malvaceae). Annals of the Missouri Botanical Garden: 439-457.
Tuskan, G. A., S. Difazio, S. Jansson, J. Bohlmann, I. Grigoriev et al., 2006 The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596-1604.
Wagenmakers, E. J., and S. Farrell, 2004 AIC model selection using Akaike weights. Psychonomic Bulletin 11: 192-196.
Wang, K., Z. Wang, F. Li, W. Ye, J. Wang et al., 2012 The draft genome of a diploid cotton Gossypium raimondii. Nature Genetics 44: 1098.
Vanneste, K., Y. Van de Peer and S. Maere, 2012 Inference of genome duplications from age distributions revisited. Molecular Biology and Evolution 30: 177-190.
Wanscher, J., 1934 The basic chromosome number of the higher plants. New Phytologist 33: 101-126.
Wendel, J. F., 2015 The wondrous cycles of polyploidy in plants. American Journal of Botany 102: 1753-1756.
Wendel, J. F., and R. C. Cronn, 2003 Polyploidy and the evolutionary history of cotton. Advances in Agronomy 78: 139.
Wendel, J. F., A. Schnabel and T. Seelanan, 1995 An unusual ribosomal DNA sequence from Gossypium gossypioides reveals ancient, cryptic, intergenomic introgression. Molecular Phylogenetics and Evolution 4: 298-313.
Wickett, N. J., S. Mirarab, N. Nguyen, T. Warnow, E. Carpenter et al., 2014 Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences USA 111: E4859-E4868.
Wilson, F. D., 1994 The genome biogeography of Hibiscus L. section Furcaria DC. Genetic Resources and Crop Evolution 41: 13-25.
Wilson, F. D., 2006 A distributional and cytological survey of the presently recognized taxa of Hibiscus section Furcaria (Malvaceae). Bonplandia: 53-62.
Wood, T. E., N. Takebayashi, M. S. Barker, I. Mayrose, P. B. Greenspoon et al., 2009 The frequency of polyploid speciation in vascular plants. Proceedings of the National Academy of Sciences USA 106: 13875-13879.
Woodhouse MR, Schnable JC, Pedersen BS, Lyons E, Lisch D, Subramaniam S, Freeling M. 2010 Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 8:e1000409.
Xie, Y., G. Wu, J. Tang, R. Luo, J. Patterson et al., 2014 SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30: 1660-1666.
Xu, Q., G. Xiong, P. Li, F. He, Y. Huang et al., 2013 Correction: Analysis of Complete Nucleotide Sequences of 12 Gossypium Chloroplast Genomes: Origin and Evolution of Allotetraploids. PloS One 8.
Zheng Li, Michael S Barker, Inferring putative ancient whole-genome duplications in the 1000 Plants (1KP) initiative: access to gene family phylogenies and age distributions, Giga Science 9:giaa004.
Zhang, Y., G.-h. Xu, X.-y. Guo and L.-j. Fan, 2005 Two ancient rounds of polyploidy in rice genome. Journal of Zhejiang University. Science. B 6: 87-90.