Methods
Animals and DNA preparation
In this study, we collected whole-blood samples from the jugular vein of 10 Bamei mutton sheep at the Xianghe Bamei mutton sheep-breeding park, Bayannur, China. These blood samples were placed in EDTA vacutainer tubes for storage. These ewes were about 3 years old and selected from among 500 sheep. Only ewes with litter size showing that they had given birth more than three times were sampled. The selected ewes were grouped into two categories based on the phenotype of litter size (monotocous sheep giving birth to only one lamb in three consecutive parities and polytocous sheep giving birth to more than two lambs in two consecutive parities). Then, genomic DNA was extracted from 200 µL of sheep blood using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), in accordance with the manufacturer’s instructions.
Genome sequencing
High-quality DNA for Illumina sequencing library construction was randomly sheared into small pieces (300–400 bp). After end-repair, “A”-tailing and ligating to Illumina sequencing adapters, 400–500 bp ligated products were amplified by ligation-mediated PCR (LM-PCR). Then, 2 × 100 bp paired-end sequencing was carried out on an Illumina HiSeq 2500 sequencer and the original data were analyzed by Illumina HiSeq Control Software.
Read processing and variant calling
NGS QC Toolkit v2.3.3 was used for quality control of the raw reads following three steps (42). First, reads with > 70% low-quality bases (score < 20) in the FASTQ files were filtered out. Second, reads containing N residues were filtered out. Third, low-quality ends (scores < 20) were trimmed. After this trimming, if the read length was < 35, the read was removed. After this quality control, the reads of each sheep were mapped to the sheep genome assembly v3.1 (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA_000298735.1_Oar_v3.1/GCA_000298735.1_Oar_v3.1_genomic.fna.gz) using BWA v0.6.2 (43). The .bam file was sorted by chromosome and duplicated reads were removed using SAMtools v0.1.19 (44).
Mapped reads of all samples were pooled for variant calling using SAMtools, with the parameters “mpileup –u –C50 –DS –q20.” The .vcf file was generated by bcftools view with the parameter “-evcgN.” Then, vcfutils.pl with minimum depth “-d 20” and maximum depth “-D 300” was used to filter raw variants. Finally, the variants were annotated by ANNOVAR (45) version 2014-11-12, in accordance with Ensembl gene annotation (Oar_v3.1).
Population genetics analysis
The samples were separated into two groups (five monotocous individuals, five polytocous individuals). The SNP densities, minor allele frequencies, and Tajima’s D of each group were calculated by VCFtools v0.1.12b (46). Only SNPs in autosomes were preserved for the phylogenetic analysis. A phylogenetic tree of all samples was generated using SNPhylo (47) version 20160204, based on the maximum likelihood method. A total of 500,000 SNPs were randomly selected for calculating the linkage disequilibrium (LD) r2 using Haploview (48) with parameters set as follows: “--missingCutoff 0.2 --dprime --minMAF 0.1.” The SNP pairs were clustered based on the physical distances of these genes. The average LD (e.g., 0–1 kb) of each group was represented by the mean r2.
Selective sweep analysis
The pooled heterozygosity Hp was calculated over 10 kb windows using the formula
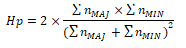
where ΣnMAJ denotes the sum of major allele frequencies in a selected window and ΣnMIN denotes the sum of minor allele frequencies(49).
FST values were calculated between monotocous individuals and polytocous individuals for individual SNPs using a method that adjusts for a small sample size (50). We averaged FST values over 50 kb sliding windows along the genome with the Bio::PopGen::PopStats Package in BioPerl (51) and Z-transformed the resultant distribution. Putative selection targets were extracted from the extreme tail of the distribution by applying a Z(FST) > 5 cut-off (24).
fastPHASE v1.4.0 was used to phase the genotypes of all samples with the parameters “-T10 –K8” (52). Then, the phased data were used to calculate the cross-population extended haplotype homozygosity (XP-EHH) value by XP-EHH (53). XP-EHH values were averaged over 50 kb sliding windows. These scores approximately followed a normal distribution; the threshold to locate putatively selected regions was two times the XP-EHH distribution standard deviation (|XP-EHH| > 2). Manhattan plots for FST and XP-EHH were generated with the R package gap.
A phylogenetic tree was generated using all variants located in these regions. Candidate genes targeted by positive selection were defined as genes overlapping with sweep regions (ZFST > 5 and |XP-EHH| > 2). GO and KEGG enrichment analyses for candidate genes were performed by DAVID 6.8 (54), and P values were corrected using the Benjamini–Hochberg method. Protein-altering mutations in these genes were listed and ranked by their single-site FST value. The protein-altering mutations of the candidate sweep gene KDM4B were localized to regions that are evolutionarily conserved among mammalian species.
Sanger sequencing validation
To confirm the SNPs detected in exons of the genes selected by sweep analysis, we selected eight SNPs from six genes and designed primers for their Sanger sequencing (Supplementary Table S1). Then, we outsourced the amplification and screening of SNPs in 15 monotocous and 14 polytocous sheep of this group to GENENODE (Wuhan, China).