In recent years, with the rapid development of modern biotechnology such as biochip and high-throughput sequencing, the increasing maturity of bioinformatics analysis, data analysis and mining of candidate genes play a leading role in the progress of diseases gradually. Bioinformatics analysis can provide fresh ideas about the study of the pathogenesis of diseases and screen for therapeutic targets. The incidence of type 2 diabetes is rapidly increasing, nevertheless, the exact pathogenesis is still unclear. Type 2 diabetes is a metabolic disease with multiple genes involved. Exploring the molecular level dysfunction, in particular, targeting key abnormal genes in islet cells of type 2 diabetes can provide efficacious analysis of differentially expressed genes and related biological functions and signaling pathways to type 2 diabetes. These are extremely important for the elucidation of the pathogenesis of type 2 diabetes.
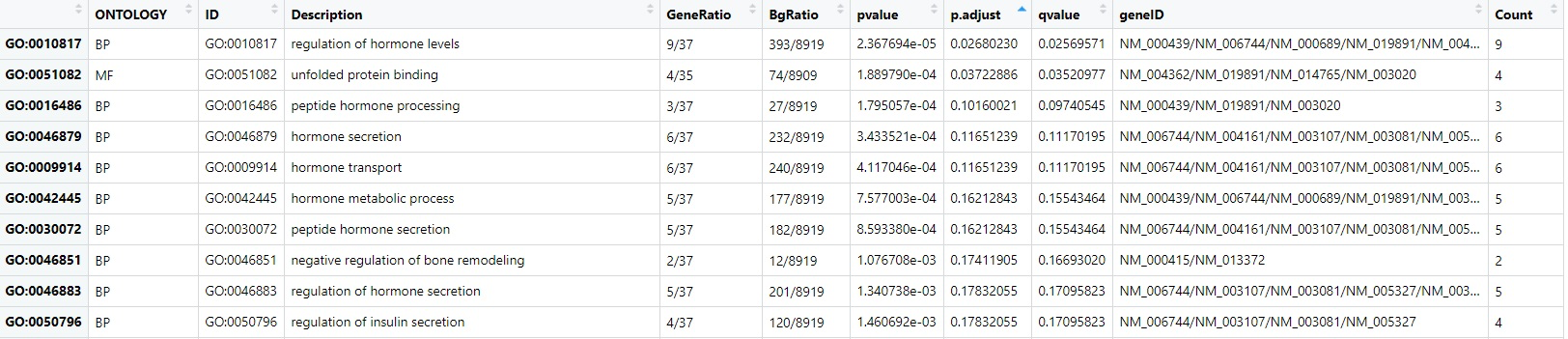
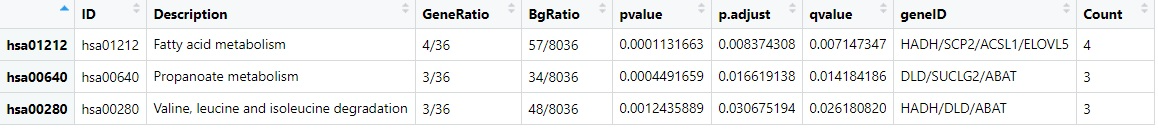
Dominguez V isolated human islets from the pancreas of 7 non-diabetics and 6 type 2 diabetic organ donors by collagenase digestion followed by density gradient purification. They performed microarray analysis to evaluate differences in the transcriptome of type 2 diabetic human islets compared to non-diabetic islet samples. The platform is GPL96[HG-U133A]Affymetrix Human Genome U133A Array. The GEO accession number is GSE25724. In our study, we extracted the expression data from GSE25724. By using the R language limma package, we screened differentially expressed genes, which may be associated with the development of type 2 diabetes. To further investigate the interactions between the DEGs, GO function, and KEGG pathway enrichment analysis were performed. The hub genes were found by the PPI network analysis. Here, we got 75 DEGs, the vast majority of which were down-regulated, only one gene was up-regulated, it was SRY (sex-determining region Y)-box 4(SOX4). The top 30 genes with the greatest differences were showed in Fig. 1. Subsequently, we performed GO and KEGG functional enrichment on these DEGs. Besides, PPI network analysis was performed on DEGs. The GO analysis indicated that the DEGs were primarily enriched in the regulation of hormone levels, hormone secretion, hormone transport, hormone metabolic processes, and regulation of insulin secretion at the level of biological processes (BP). The DEGs were primarily enriched in unfolded protein response at the molecular functional level(MF). KEGG pathway enrichment analysis showed that DEGs were markedly enriched in fatty acid metabolism pathway, propionate metabolism pathway, degradation pathway of valine, leucine, and isoleucine.
Increased circulating lipid levels and metabolic alterations in fatty acid metabolic pathway dysfunction and intracellular signaling have turned out to be associated with insulin resistance in the muscle and liver of diabetic patients[2]. Imbalance in fatty acid metabolism can lead to impaired GSIS(glucose-stimulated insulin secretion) with concomitant oxidative and metabolic stress, endoplasmic reticulum stress, and numerous pro-apoptotic signals, all of which lead to a decrease of β-cell survival[3]. Propionate inhibits hepatic glucose gluconeogenesis via the G protein-coupled receptor 43/AMP-activated protein kinase(GPR43/AMPK) signaling pathway[4]. The plasma levels of branched-chain amino acids(valine, leucine, isoleucine) increase in conditions related to insulin resistance, such as obesity and diabetes[5]. Higher circulating level of branched-chain amino acids is strongly linked to a higher risk of type 2 diabetes[6–7]. Experimental studies have found that impairment of the adaptive unfolded protein response in mouse β-cells leads to reduce transportation from endoplasmic reticulum to Golgi protein and further increase β-cell death[8]. Our findings were generally consistent with those papers.
A PPI network of DEGs was performed by using the STRING database and Cytoscape software. SCG5, SNAP25, SCP2, CPE, and PCSK1 were the hub genes. SCG5 encodes a secretory chaperone that prevents the aggregation of other secret proteins, including those associated with neurodegenerative and metabolic diseases. It has been mostly studied for its role in the transportation and activation of the prohormone convertase 2. SCG5 acts as a molecular chaperone for kexin2 proprotein convertase subtilisin/prohormone convertase 2 (PCSK2/PC2), preventing its premature activation in the regulated secretory pathway. SCG5 binds to inactivated PCSK2 in the endoplasmic reticulum, facilitates its transport from there to later compartments of the secretory pathway where it is proteolytically matured and activated. It is found that the changes of type 2 diabetic phenotype in GK rats may be caused by the accumulation of multiple genetic variants, including the SCG5 gene, and the mutated genes may affect biological functions including adipocytokine signaling, glycerolipid metabolism, PPAR signaling, T cell receptor signaling, and insulin signaling pathways [9]. The present study found that the SCG5 gene was mainly enriched in unfolded protein binding (GO:0051082), which might be linked to hormone preprocessing and insulin secretion.
SNAP25 is interconnected with proteins that participate in vesicle docking and membrane fusion.SNAP25 regulates plasma membrane recirculation through its interaction with centromere protein F (CENPF).SNAP25 also modulates the gating characteristics of the delayed rectifier voltage-dependent potassium channel, potassium voltage-gated channel, Shab-related subfamily, member 1(KCNB1) in pancreatic beta cells. Studies have evaluated the possible role of SNAP25 polymorphisms in T2DM, suggesting that the minor SNAP25 rs363050 (G) allele, which results in a reduced SNAP25 expression is associated with altered glycemic parameters in patients with T2DM, possibly because of reduced functionality in the exocytotic machinery leading to the suboptimal release of insulin[10]. Tao Liang[11]found that SNAP23 is the ubiquitous SNAP25 isoform that mediates secretion in non-neuronal cells, similar to SNAP25 in neurons. Pancreatic islet β cells contain an abundance of both SNAP25 and SNAP23. SNAP23 depletion promotes SNAP25 to bind calcium channels more quickly and longer where granule fusion occurs to increase exocytosis efficiency. In this study, we found that the SNAP25 gene was mainly enriched in hormone transport (GO:0009914), which might be linked to insulin cytokinesis and exocrine secretion.
SCP2, a non-specific lipid-transport protein; mediates the transfer of all common phospholipids, cholesterol, and gangliosides between cell membranes. SCP2 may play a role in the regulation of steroidogenesis. It was found that SCP2 protein levels were decreased significantly in severely hypercholesterolemic diabetic animals. This differential expression of sterol carrier proteins SCP2 may accompany diabetic dyslipidemia, which should be considered a potential contributing mechanism through which cholesterol metabolism may be altered in diabetes[12]. Our study found that the SCP2 gene was enriched mainly in coenzyme binding (GO:0050662), which was involved in disorders of diabetic lipid metabolism.
CPE encodes a member of the M14 family of metallo-carboxypeptidases. It is a categorical receptor that directs hormone precursors into regulatory secretory pathways. It also serves as a hormone precursor processing enzyme in neural/endocrine cells, removing dibasic acid residues from the C terminus of peptide hormone precursors after initial endonuclease cleavage. Carboxypeptidase E is a peptide processing enzyme involved in the cleavage of numerous peptide precursors, including neuropeptides and hormones associated with appetite control and glucose metabolism including proinsulin. Diseases associated with CPE contain hyperinsulinemia and insulinoma. CPE is involved in the biosynthesis of various neuropeptides and peptide hormones in endocrine tissues and the nervous system. Loss of normal CPE leads to various disorders, containing diabetes, hyperinsulinemia, low bone mineral density, and deficits in learning and memory[13]. Truncating mutations in the CPE gene have been shown to cause morbid obesity, intellectual disability, abnormal glucose homeostasis, and hypogonadotrophic hypogonadism, it reveals the importance of CPE in the regulation of body weight and metabolism, and brain and reproductive function in humans[14]. GO annotations associated with this gene include cell adhesion molecule binding, carboxypeptidase activity, and peptide hormone processing(GO00016486).
PCSK1 also known as neuroendocrine convertase 1, encodes a member of the Bacillus subtilisin-like preprotein convertase family, which have the capacity to regulate secretory pathways or to be a component of one of the branchings, proteases that process protein and peptide precursors. PCSK1 is involved in the processing of hormone and other protein precursors at sites comprised of pairs of basic amino acid residues. Substrates include proopiomelanocortin (POMC), renin, enkephalin, dynorphin, somatostatin, and insulin. The universal genetic variants rs6232 and rs6235 within PCSK1 are found to determine glucose-stimulated proinsulin conversion, but not insulin secretion. Besides, rs6232 influences glucose homeostasis and insulin sensitivity independently of Body Mass Index (BMI) and proinsulin concentrations[15]. Rona JS et al [16]identify nine genetic variants associated with fasting insulinogen, including PCSK1, which is associated with the glucose homeostasis and T2DM development in humans and argues against a direct role of proinsulin in coronary artery disease pathogenesis. Mayumi Enya[17]found that the genetic variant of PCSK1 may influence glucose homeostasis by altering insulin resistance independently of BMI, incretin level, or proinsulin conversion, and may be associated with the occurrence of type 2 diabetes in Japanese. In our study, we found that the PCSK1 gene was mainly enriched in regulating hormone levels (GO:0010817). it was concluded that the gene is related to the regulation of hormone and glucose homeostasis in diabetes mellitus.