Protein phosphorylation acts as an essential on/off switch in many cellular signalling pathways, regulating protein function. This has led to ongoing interest in targeting kinases for therapeutic intervention. Computer-aided drug discovery has been proven a useful and cost-effective approach for facilitating prioritisation and enrichment of screening libraries. Limited effort, however, has been devoted to developing and tailoring in silico tools to assist the development of kinase inhibitors and providing relevant insights on what makes potent inhibitors. To fill this gap, here we developed kinCSM, an integrative computational tool capable of accurately identifying potent cyclin-dependent kinase 2 (CDK2) inhibitors, quantitatively predicting CDK2 ligand-kinase inhibition constants (pKi) and classify inhibition modes without kinase information. kinCSM predictive models were built using supervised learning and leveraged the concept of graph-based signatures to capture both physicochemical properties and geometry properties of small molecules. CDK2 inhibitors were accurately identified with Matthew’s Correlation Coefficients of up to 0.74, and inhibition constants predicted with Pearson’s correlation of up to 0.76, both with consistent performances of 0.66 and 0.68 on non-redundant blind tests, respectively. kinCSM was also able to identify the potential type of inhibition for a given molecule, achieving Matthew’s Correlation Coefficient of up to 0.80 on cross-validation and 0.73 on blind test. Analysing the molecular composition of kinase inhibitors revealed enriched chemical fragments in potent CDK2 inhibitors and different types of inhibitors, which provides insights into the molecular mechanisms behind ligand-kinase interactions. We believe kinCSM will be an invaluable tool to guide future kinase drug discovery. To aid the fast and accurate screening of potent CDK2 kinase inhibitors, we made kinCSM freely available online at http://biosig.unimelb.edu.au/kin_csm/.
Research Article
kinCSM: using graph-based signatures to predict small molecule CDK2 kinase inhibitors
https://doi.org/10.21203/rs.3.rs-669465/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Kinase inhibitors
small molecules
machine learning
bioactivity prediction
graph-based signatures
CDK2 inhibitors
The human genome encodes more than 500 protein kinases which catalyze the process of transferring phosphate groups1. Kinases are important in many cellular signalling processes, including cell growth, proliferation, apoptosis, and metabolism2, with abnormal kinase regulation leading to a range of diseases, including cancer3. It has been proposed that over a third of human protein functions are regulated by phosphorylation, making kinases attractive targets for therapeutic interventions via inhibition or modulation.
Developing kinase inhibitors via the traditional drug development process, however, is a time-consuming and costly endeavour. To date, only 52 inhibitors have been approved by the U.S. Food and Drug Administration (FDA), targeting a small fraction of human kinases4. While the traditional in vivo essays for hit discovery are challenging and usually present low hit-rates, data availability emerging from these efforts has led to developments in virtual screening, a time- and cost-effective approach to enable improvement in discovery rates and prioritisation of compounds5. One approach that has successfully leveraged this data has been quantitative structure − activity relationship (QSAR)6 analyses have been playing an important role in drug discovery efforts7. Balachandar et al. identified potent inhibitors targeting 8 kinases by using deep learning models8, and Govinda et al. predicted drug-kinase inhibition constant (pKi) for a wide range of kinases9. Additionally, Miljković et al. classified different types of inhibition based on binding modes by considering a ligand-based approach10. Although these models represent a significant contribution to the field, they presented poor performance and generalisation capabilities, and provided limited biological insight into what physicochemical properties are required to the design of new potent kinase inhibitors, for different binding modes.
Cyclin-dependent kinases (CDKs) within the family of Ser/Thr kinases can drive the cell cycle propagation upon bindings to cyclins. They have become popular chemotherapeutic targets for different types of cancers. While a number of studies have been focused on CDK4/6 inhibitors to mediate tumour cell cycle arrest, CDK2 can also be a promising target to overcome drug resistance to CDK4/6 inhibitors11. To our knowledge, there has been no freely accessible tool dedicated to predict the potency of CDK2-targeting small molecules and their binding modes.
We have previously shown that the concept of graph-based signatures used to model both protein and small molecule structures12, 13, 14, 15, 16, 17, capturing both geometry and physicochemical properties 18, 19, 20, 21. Leveraging this concept, we developed kinCSM (Fig. 1), a new predictive tool dedicated to identify potent CDK2 inhibitors. The method has three different predictive capabilities. Firstly, it can accurately identify potential CDK2 inhibitors. Secondly, it can quantitatively measure potency by predicting the inhibition constant (pKi), allowing compounds to be ranked and prioritised. Finally, it also enables identification of the mode of inhibition. We show kinCSM performs as well as or better than similar methods and can generate biological insights into what makes potent CDK2 inhibitors.
Associating molecular properties with CDK2 inhibition
By analyzing the general physicochemical properties of compounds, we found no strong correlation between independent molecular features and the inhibition constant, pKi (Pearson’s correlation coefficient of up to 0.21). Across our datasets, both CDK2 inhibitors and non-inhibitors generally followed Lipinski's rule of five (RO5)22 and Veber's Rule23, reflecting an intrinsic bias in the screening libraries routinely used. Most of the active molecules evaluated had no more than 10 hydrogen bond acceptors, less than 5 hydrogen bond donors, octanol-water partition coefficient (log P) less than 5, no more than 10 rotatable bonds, polar surface area (TPSA) less than 140 Å2 (Figure S1).
Despite a modest correlation between inhibition strength and drug-likeness properties, some physicochemical properties did distinguish between CDK2 inhibitors and non-inhibitors. Potent CDK2 inhibitors had a lower log P (Figure S1 C) (p-value < 0.001, using a two-sample Kolmogorov − Smirnov test), indicating they are more hydrophilic and are more likely to be distributed in aqueous regions such as blood serum. Consistent with this observation, inhibitors also had a larger TPSA (Figure S1 E) (p-value < 0.001) compared to non-inhibitors, reflecting a potential to establish more interactions with kinases.
Molecular substructure mining
To further our understanding of the chemical landscape of known kinase inhibitors, we used molecular substructure mining to identify enriched chemical groups. Using the Molecular Substructure Miner (MoSS)24, we found two chemical fragments, sulfanilamide (16.2% support) and 2-(N-Anilino)pyrimidine (10.1% support), that occurred more frequently in CDK2 inhibitors compared to non-inhibitors (Figure S2). Atoms in these enriched groups include hydrogen bond donors and acceptors, and the two negatively charged oxygens in sulfonamide that can form electrostatic interactions with positively charged amino acids in the kinase. Additionally, the ring structures in the fragments can mimic the adenine component of ATP, important for competitive inhibitors.
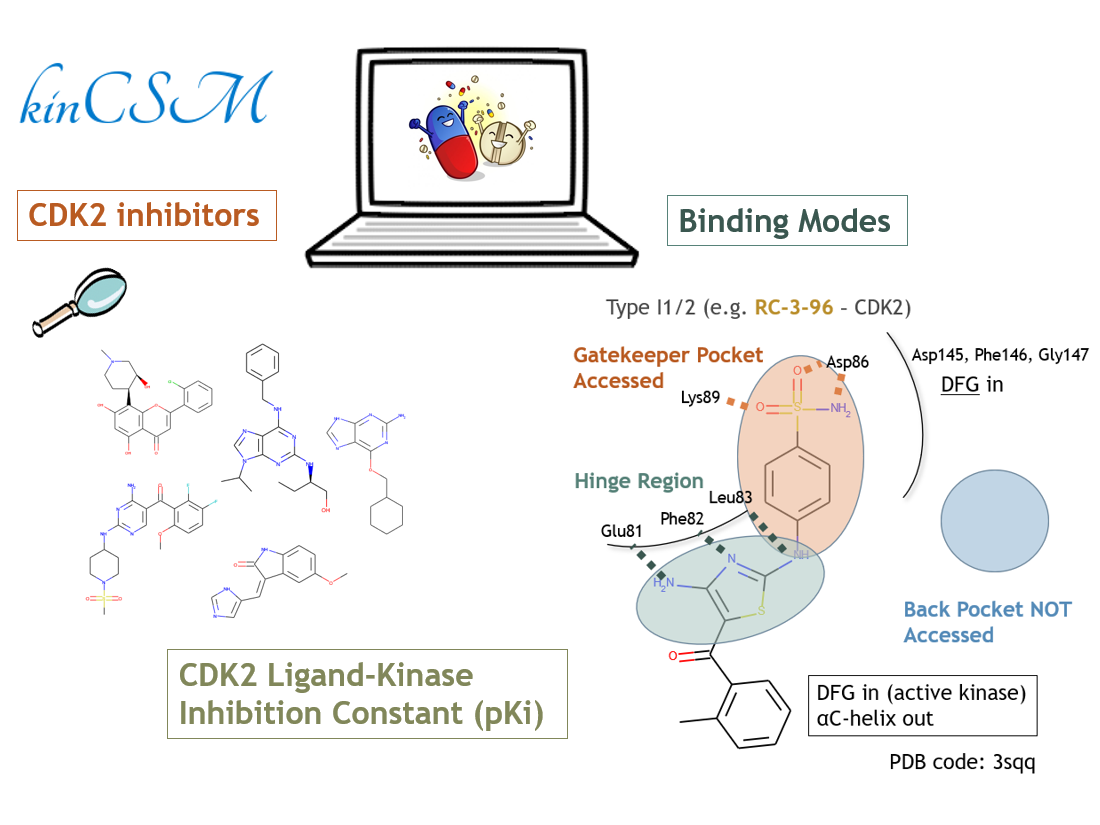
Two enriched fragments, sulfanilamide (10.7% support) and 4-Amino-1,3-thiazole-5-carbaldehyde (8.6% support) were found in type I1/2 inhibitors (Fig. 2A). By searching molecules containing both fragments in the Protein Data Bank25, we found that both substructures can form hydrogen bonds with gatekeeper residues and the hinge region, respectively. While they occur together in type I1/2 inhibitors less frequently (4.1% support), these substructures never appear together in other types of inhibitors. This may suggest a distinctive binding mode of type I1/2 inhibitors, the coordinated interactions with the gatekeeper pocket and the hinge region caused by the αC-helix out and DFG in kinase conformation.
The enriched substructure (24.2% support) in type II inhibitors is composed of a 1-Phenylurea connected to a ring (Fig. 2B). The odds ratio is 64.7 compared to type I, and 41.6 compared to type I1/2, indicating confident enrichment. Urea can form a hydrogen bond donor-acceptor pair with the αC-helix and DFG motif, consistent with experimentally solved structures. The nitrogen atoms can establish hydrogen bonds with the glutamate side chain, which is conserved in αC-helix, while the carbonyl group can establish a hydrogen bond with the backbone amide of the aspartate in the DFG-motif. The benzene ring close to the donor nitrogen can form aromatic interactions with the gatekeeper residue in the kinase and a hydrophobic moiety (at the top right corner in Fig. 2B) accommodates it in the back pocket. Accordingly, the urea acts as a bridge between the two ring structures and the two pockets exposed by the DFG out and αC-helix out kinase conformation.
Substructure enrichment for type I and allosteric inhibitors was not thoroughly analysed. Type I inhibitors form stronger interactions with the hinge region similar to ATP, without having access to the back pocket and gatekeeper area (Fig. 2C). As both type I1/2 and type II inhibitors share common substructures capable of occupying the ATP binding site, no substructure was found exclusively in type I inhibitors. Additionally, the limited sample size for allosteric inhibitors (32 in 10-fold cross-validation, 15 in blind test) did not allow for an unbiased enrichment analysis.
Identifying CDK2 inhibitors
Our predictive model was trained using different supervised learning algorithms. The best performing algorithm, Extra Tree Classifier (M5P) with 23 features (identified via feature selection), was chosen. Table 1 shows the overall model performance. Although the dataset used is relatively unbalanced (595 non-inhibitors, 1040 inhibitors), the model still achieved high and consistent Matthew’s Correlation Coefficients (MCCs) on both 10-fold cross-validation (0.74) and independent blind test set (0.66). F1 score (0.91 on cross-validation, 0.88 on blind test) and AUC (0.86 on cross-validation, 0.84 on blind test) also demonstrated model robustness (Fig. 3). The performance metrics obtained via rigorous internal and external validation suggest potent CDK2 inhibitors can be correctly identified.
| MCC | F1 | AUC | |
|---|---|---|---|
| 10-fold CV | 0.74 | 0.91 | 0.86 |
| blind test | 0.66 | 0.88 | 0.84 |
To shed light into properties that can explain differences between CDK2 inhibitors and non-inhibitors, we conducted a two-sample Kolmogorov-Smirnov test on the feature set. Figure S3 A shows the top three features with the smallest p-values. Inhibitors tend to have higher partial charges and van der Waals surface area contributions (PEOE_VSA12 attribute), a higher frequency of sulfonamides, and more hydrogen bond donors (p-values < 0.001). These characteristics reveal different non-covalent interactions between enriched substructures (sulfanilamide and 2-(N-Anilino)pyrimidine) and CDK2, including electrostatic interactions, hydrogen bonds, and van der Waals forces which can stabilize favour inhibitor binding.
Compared to the deep learning models developed by Balachandar et al. on the same dataset, our classical machine learning algorithm has competitive performance. On the blind test, we achieved an AUC of 0.84, whereas Balachandar et al. achieved an AUC of 0.738. lthough the performance results are not directly comparable since the training and test set splits are different, our model does demonstrate satisfactory generalization. The small score difference between 10-fold cross-validation (0.86) and blind test (0.84) provides further confidence in model robustness. Additionally, by investigating both the significant features and enriched substructures, we inferred discriminative physicochemical properties of potent inhibitors and discussed their biological significance. In contrast, no relevant biochemical insight was drawn from previous works2, as features were encoded as bit strings to accommodate deep learning architectures, which are not explainable. Therefore, our model does not only have competitive prediction performance but also contributes to the detection of novel scaffolds among potent inhibitors and shed light into their potential mode of action.
Predicting CDK2 ligand-kinase inhibition constant (pKi)
By predicting the pKi values of small molecules, the inhibition strength can be quantified. A Random Forest Regressor (RF) with 22 features was trained and validated. Table 2 shows the overall model performance. We obtained a Pearson’s correlation coefficient of 0.76 (RMSE of 0.62) on 10-fold cross-validation, and 0.68 (RMSE of 0.65) on an independent blind test set. The consistent performance between internal and external validation indicates model generalisation. After removing 10% of outliers, Pearson’s correlation coefficients increased to 0.87 on cross-validation and 0.78 on blind test (Fig. 4). Here, no enriched substructures were observed exclusively in outlier molecules, indicating their structural diversity.
| Pearson | Spearman | Kendall | MSE | RMSE | |
|---|---|---|---|---|---|
| 10-fold CV | 0.76 | 0.71 | 0.56 | 0.39 | 0.62 |
| blind test | 0.68 | 0.59 | 0.45 | 0.43 | 0.65 |
By dividing the molecules into two groups with the cut-off pKi value of 6, we were able to compare the physicochemical differences between the defined potent inhibitors (pKi ≥ 6) and non-inhibitors (pKi < 6) in cell-based assays using the two-sample Kolmogorov-Smirnov test. Figure S3B depicts three significant features (p-values < 0.001) discriminating molecules with a high binding affinity (pKi ≥ 6). These features were consistent with those identified previously.
While the regression model with the pKi cut-off could also potentially be useful for classification purposes, in general continuous labels can have higher variance compared to discrete classes, and may lead to poor classification performance.
To test this assumption, we converted the predicted and the true pKi values to inhibitor and non-inhibitor classes. As expected, after the label transformation, the model achieved lower MCC (0.64 on cross-validation, 0.57 on blind test) compared to our dedicated CDK2 inhibitor classification model (0.74 on cross-validation, 0.66 on blind test). Accordingly, rather than being a substitute for the classification model, our regression model can serve as a tool to quantify and rank inhibition strength in addition to inhibitor identification.
Classifying different types of kinase inhibitors
The dataset for classification of inhibitor type is highly unbalanced (1425 type I, 394 type I1/2, 190 type II, and 47 allosteric inhibitors), which significantly increases the challenges of identifying the minority classes. However, our model was able to distinguish type II inhibitors from type I1/2 inhibitors, despite their smaller sample sizes. As shown in Table 3, the type I1/2 versus type II classifier achieved MCCs of 0.80 on cross-validation and 0.73 on blind test sets. Additionally, it also achieved the highest AUC with 0.91 on the blind test set (Figure S4). The method has also identified allosteric inhibitors effectively, with a MCC of 0.68 on cross-validation and 0.63 on blind test.
| Classifier | Metric | kinCSM cross validation | kinCSM blind test | Miljkovic et al.10blind test |
|---|---|---|---|---|
| Type I versus II | F1 | 0.73 | 0.64 | 0.71 (± 0.03) |
| BACC | 0.80 | 0.74 | 0.78 (± 0.02) | |
| MCC | 0.73 | 0.65 | 0.70 (± 0.04) | |
| Type I versus I1/2 | F1 | 0.54 | 0.43 | 0.58 (± 0.04) |
| BACC | 0.69 | 0.64 | 0.74 (± 0.02) | |
| MCC | 0.50 | 0.41 | 0.47 (± 0.05) | |
| Type I1/2 versus II | F1 | 0.87 | 0.82 | 0.77 (± 0.03) |
| BACC | 0.90 | 0.88 | 0.82 (± 0.02) | |
| MCC | 0.80 | 0.73 | 0.69 (± 0.03) | |
| Allosteric or not | F1 | 0.64 | 0.57 | 0.36 (± 0.18) |
| BACC | 0.73 | 0.70 | 0.63 (± 0.07) | |
| MCC | 0.68 | 0.63 | 0.48 (± 0.09) |
Compared to the best machine learning model developed by Miljković et al.10 was validated on a randomly generated external blind test set, our model achieved higher MCCs in identifying allosteric inhibitors and distinguishing type I1/2 and II inhibitors even when the blind test set presents low similarity with the training set (Table 3). This means our model has a better generalisation for unseen data when the sample size is limited and unbalanced.
Another challenge for this task was to do with the molecular structures of the three ATP competitive inhibitor types, which can be modelled as a continuum instead of distinct categories as the kinase conformation they bind changes in a stepwise manner26. Type I inhibitors bind to the DFG-in, αC-helix in conformation, then the movement of αC-helix (DFG-in, αC-helix out) allows binding of type I1/2 inhibitors, and lastly, the DFG-out, αC-helix out conformation is recognized by type II inhibitors. Two selected machine learning features (fluorine and hydrophobe counts) demonstrate this continuum (Figure S5 A and B). The distributions of type I1/2 inhibitors can be visualized as a mixture of type I and type II inhibitors, biased towards type I. This may suggest that the shared substructures between types of binding mode can affect model performance.
Being positioned in the middle of the continuum, type I1/2 becomes the most challenging class, even though it has an adequate sample size. The type I versus type I1/2 classifier achieved the lowest performance (MCC of 0.50 on cross-validation, and 0.41 on blind test, shown in Table 3). After integrating the prediction outcomes from the four binary classification models, a large proportion of type I1/2 inhibitors were wrongly classified as type I inhibitors (Figure S6). One possible reason is that type I1/2 inhibitors share a larger proportion of common substructures with type I inhibitors in comparison with type II inhibitors. Although type I1/2 inhibitors can form interactions with residues in the gatekeeper pocket, making them distinguishable from type I, this characteristic may not be captured by our model. Rather, their strong affinity with the hinge region lead to similar physicochemical properties (e.g., low lop P as shown in Figure S5 C) as type I inhibitors. Nevertheless, our model does capture features capable of distinguishing type I1/2 inhibitors from others (e.g., higher frequency of nitrogen-containing functional groups attached to aromatics, as shown in Figure S5 D - Welch two-sample t-test p-values < 0.001 compared to type I and II).
Although type II inhibitors have a distinctive characteristic (back pocket access), larger sample size still causes biased predictions towards type I inhibitors (Figure S6). The type I versus II classifier achieved MCC of 0.73 and 0.65 for 10-fold cross-validation and blind test respectively (Table 3). However, insightful features were captured by our model. Type II inhibitors have higher log P (p-values < 0.001 compared to type I and I1/2) as shown in Figure S5 C, which means they are more hydrophobic. This is caused by their special interactions with the kinase hydrophobic back pocket. Additionally, fluorine and urea occur more frequently in type II inhibitors (p-values < 0.001, Figure S5 A and E). This may suggest both of them can contribute with interactions with the back pocket.
kinCSM Web Server
kinCSM has been made freely available through an easy-to-use web interface at http://biosig.unimelb.edu.au/kin_csm/. Users can identify CDK2 inhibitors, predict CDK2 pKi and possible binding modes irrespective of kinases by providing a single molecule or a list of molecules as SMILES strings (Fig. 5).
Here we developed kinCSM, the first predictive tool to identify CDK2 inhibitors, predict CDK2 Ligand-Kinase Inhibition Constant (pKi), and classify different types of inhibitors in a single resource. This tool can be used to study both the binding affinity and binding modes of kinase inhibitors.
Using the concept of graph-based signatures, our model not only achieved high prediction performance but also inferred distinctive physicochemical properties that are supported by substructure mining. We have made the kinCSM webserver freely available at http://biosig.unimelb.edu.au/kin_csm/.
We anticipate further model optimization by generating substructure descriptors and oversampling the minor class in the future. The model can also be trained to target different kinases for inhibitor selectivity studies. This may create extra value for drug development. We believe kinCSM would be a useful tool for accelerating kinase inhibitor drug screening and improving hit rates.
Data sets
Molecules were curated from three different literature sources8, 9, 10 for the three aims, and converted into SMILES strings. The label distributions of the three datasets are all unbalanced to some extent. Dataset 1 has more CDK2 inhibitors (63.6%) than non-inhibitors (36.4%), and the pKi distribution in Dataset 2 has a peak at around 5. Additionally, most of the inhibitors discovered so far are type I, and only a few allosteric inhibitors have been developed. This leads to the highly unbalanced dataset 3 (1425 type I, 394 type I1/2, 190 type II, and 47 allosteric inhibitors) for inhibitor type classification. All data sets used in this study are available at http://biosig.unimelb.edu.au/kin_csm/data.
The datasets were split into low-redundancy training (70%) and blind test (30%). We ensured the molecules in the training and blind test sets have similar label distribution but are in different similarity clusters. The clusters were formed using the rdkit.ML.Cluster.Butina module in the cheminformatics toolkit RDKit27 according to the TanimotoSimilarity28. The similarity thresholds were adjusted to ensure that half of the molecules in the dataset are singletons, and the other half have at least one neighbor within their clusters.
Graph-based signatures and feature selection
Molecular features for machine learning were extracted from SMILES strings as done previously18, 19, 20. This approach has been successfully used on a variety of datasets to predict pharmacokinetic properties, including both classification (with categorical labels) and regression (with continuous labels). It generates both physicochemical features and graph-based signatures, making it an effective way to represent molecules’ properties.
The graph-based signatures are distance patterns that are generated iteratively by the Cutoff Scanning Matrix (CSM) algorithm13, 14, 29. Molecules are modelled as a graph in an undirected and unweighted way, where atoms are represented as nodes, and bonds are represented as edges. Additionally, all atoms are labelled with pharmacophores (including Acceptor, Donor, PosIonizable, NegIonizable, Aromatic, and Hydrophobe) as shown in the bottom left panel of Figure S7. While scanning through the whole molecular graph, the distances between pharmacophore pairs are captured as a cumulative distribution using all-pairs shortest paths (bottom right panel of Figure S7). This information can add extra values to the feature space, and therefore facilitate quantitative structure − activity relationship (QSAR) investigation.
Model selection and evaluation
Different machine learning models were trained and assessed under 10-fold cross-validation. We then evaluated the trained models on the blind test set and compared the performance of the machine learning methods.
Specifically, in this study, we have compared the performance of the following popular machine learning techniques using the python Scikit-learn library30: random forest, extra trees, multilayer perceptrons, support vector machines, and k-nearest neighbours. Our evaluation result suggests that tree-based methods lead to the highest performance for the regressor and most of the classifiers, except multilayer perceptron, is the best method for type I1/2 versus type II classifiers.
Finally, the model performance was further evaluated by different metrics. MCC, F1 score and AUC for classification, Pearson’s correlation coefficient (r), mean squared error (MSE) and root mean squared error (RMSE) for regression.
A bottom-up greedy feature selection method was used according to the Matthew’s Correlation Coefficient (MCC) for classification, and Pearson’s Correlation Coefficient (r) for regression, to simplify models and reduce noise.
Substructure mining
The SMILES strings were input into the Molecular Substructure Miner (MoSS)24 to investigate substructure enrichment. We searched enriched substructures in a focused group of molecules (inhibitors) compared to a complementary set (non-inhibitors). Discriminative fragments were found in CDK2 inhibitors compared to non-inhibitors, and also for different types of kinase inhibitors in a pair-wise manner. These substructures and patterns can further validate the features learned by our models, and also improve their overall interpretability. Finally, we studied the kinase-ligand interaction patterns by searching molecules enriched with these substructures in the Protein Data Bank (PDB)25.
The odds ratios for substructure enrichment were calculated based on the contingency tables obtained from control studies. They can quantify the association between enriched fragments and the inhibitors. Table S1 shows an example of the contingency table for the top left fragment (in the blue box) in Figure S2. The odds ratio was calculated as:

Odds ratios greater than one for both of the fragments demonstrate their confident enrichments in inhibitors.
Web server development
The web server front end was developed using Bootstrap framework version 3.3.7, and the back end was based on Python 2.7 via the Flask framework version 0.12.3 on a Linux server running Apache.
Availability of data and materials
All data is freely available at: http://biosig.unimelb.edu.au/kin_csm/data.
Competing interests
The authors declare no competing interests.
Funding
R.A is funded by a PhD scholarship from the Kingdom of Saudi Arabia. This work was supported in part by the Medical Research Council (MR/M026302/1 to D.B.A. and D.E.V.P.); the National Health and Medical Research Council of Australia (GNT1174405 to D.B.A.), the Wellcome Trust (093167/Z/10/Z), and the Victorian Government’s Operational Infrastructure Support Program. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
- Duong-Ly KC, Peterson JR. The human kinome and kinase inhibition. Curr Protoc Pharmacol Chap. 2, Unit2 9 (2013).
- Miljkovic, F., Rodriguez-Perez, R. & Bajorath, J. Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes. J Med Chem, 63, 8738–8748 (2020).
- Pandey, K. et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int J Cancer, 145, 1179–1188 (2019).
- Myung, Y., Pires, D. E. V. & Ascher, D. B. mmCSM-AB: guiding rational antibody engineering through multiple point mutations. Nucleic Acids Res, 48, W125–W131 (2020).
- Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures., 30, 335–342 (2014).
- Pires, D. E., Blundell, T. L. & Ascher, D. B. mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep, 6, 29575 (2016).
- Pires, D. E. V., Rodrigues, C. H. M. & Ascher, D. B. mCSM-membrane: predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res, 48, W147–W153 (2020).
- Rodrigues, C. H. M., Pires, D. E. V. & Ascher, D. B. mmCSM-PPI: predicting the effects of multiple point mutations on protein-protein interactions.Nucleic Acids Res, (2021).
- Rodrigues, C. H. M., Pires, D. E. V. & Ascher, D. B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci, 30, 60–69 (2021).
- Pires, D. E. V. & Ascher, D. B. mycoCSM: Using Graph-Based Signatures to Identify Safe Potent Hits against Mycobacteria. J Chem Inf Model, 60, 3450–3456 (2020).
- Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem, 58, 4066–4072 (2015).
- Pires, D. E. V., Stubbs, K. A., Mylne, J. S. & Ascher, D. B. Designing safe and potent herbicides with the cropCSM online resource. bioRxiv, 2020.2011.2001.364240(2020).
- Kaminskas, L. M., Pires, D. E. V. & Ascher, D. B. dendPoint: a web resource for dendrimer pharmacokinetics investigation and prediction. Sci Rep, 9, 15465 (2019).
- Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 46, 3–26 (2001).
- Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem, 45, 2615–2623 (2002).
- Borgelt, C., Meinl, T. & Berthold, M. MoSS: a program for molecular substructure mining. In: Proceedings of the 1st international workshop on open source data mining: frequent pattern mining implementations (ed^(eds). Association for Computing Machinery(2005).
- Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res, 28, 235–242 (2000).
- Roskoski, R. Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res, 103, 26–48 (2016).
- Landrum, G. RDKit: Open-source cheminformatics(2006).
- Butina, D. Unsupervised Data Base Clustering Based on Daylight's Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. Journal of Chemical Information and Computer Sciences, 39, 747–750 (1999).
- Pires, D. E. & Ascher, D. B. CSM-lig: a web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res, 44, W557–561 (2016).
- Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res, 12, 2825–2830 (2011).
- Schonbrunn, E. et al. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J Med Chem, 56, 3768–3782 (2013).
- Alevy, Y. G. et al. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest, 122, 4555–4568 (2012).
- Metz, J. T. et al. Navigating the kinome. Nat Chem Biol, 7, 200–202 (2011).
- 26. Roskoski R, Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res 103, 26–48 (2016).
- Landrum G. RDKit: Open-source cheminformatics. (2006).
- Butina D. Unsupervised Data Base Clustering Based on Daylight's Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. Journal of Chemical Information and Computer Sciences 39, 747–750 (1999).
- Pires DE, Ascher DB. CSM-lig: a web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res 44, W557-561 (2016).
- Pedregosa F, et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res 12, 2825–2830 (2011).
- Schonbrunn E, et al. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J Med Chem 56, 3768–3782 (2013).
- Alevy YG, et al. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest 122, 4555–4568 (2012).
- Metz JT, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ. Navigating the kinome. Nat Chem Biol 7, 200–202 (2011).
No competing interests reported.
- TableofContentsgraphic.png
Table of Contents graphic
- kinCSMsupplementary.docx
{kind=link}