Case Reports and Results
Case 1
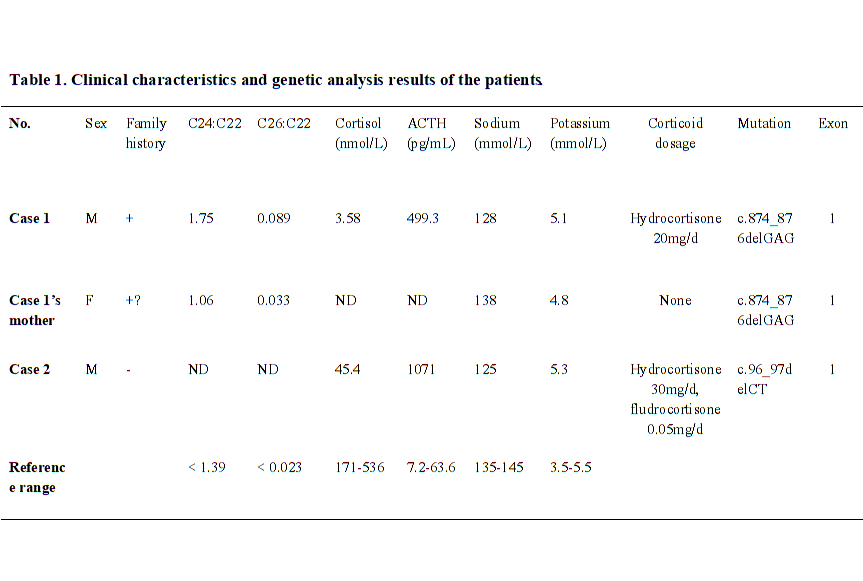
A 27-year old male was presented in hospital with weakness, fatigue, poor academic performance, exhibiting skin and mucous membrane pigmentation, especially in areolae, joints and axilla for two decades. The cortisol at 8am was reduced 3.58 nmol/L (range 171–536 nmol/L), and adrenocorticotropic hormone (ACTH) was increased 499.30 pg/mL (range 7.2–63.6 pg/mL). After diagnosed as primary adrenal insufficiency (Addison’s disease), he was administrated with glucocorticoid replacement (hydrocortisone 20 mg per day) for one year, and the skin pigmentation and weakness were improved. In recently half a year, the patient acted mild cognitive impairment, slurred speech, ataxia, and rapidly progressed as walking instability, incontinence, and paroxysmal blurring vision. Physical examination detected scanty scalp hair, V−grade muscle strength, hypomyotonia, tendon hyperreflexia (+++), positive Barbinski’s sign and Gordon’s sign. The light and deep reflex were normal. The heel-to-knee test, alternating movement test, and Romberg’s test were negative. The respiratory, cardiovascular, and abdominal examinations were unremarkable. Brain MRI showed symmetrical distribution of hyperintensity long T1 and long T2 signal in of bilateral splenium of corpus callosum, and widened fissures and sulci (Fig. 1A). Serum VLCFA levels were significantly increased (Table 1). Genetic sequencing detected heterozygous mutation of c.874_876delGAG in ABCD1 gene Exon 1, leading to the deletion of translation product glutamate and accumulation of VLCFAs. Sanger sequencing of ABCD1 gene was performed on his family members (Fig. 2A and 2B). His mother was confirmed as the heterozygous point mutation for the same variant. The serum VLCFA levels of his mother were elevated (Table 1). Until now, his mother did not show manifestations of cerebral disorders, myeleterosis, or adrenal insufficiency. His maternal grandmother died at her 55-year-old due to encephalopathy. However, family members were unable to provide valid information, and no blood samples could be obtained for testing. Other siblings of his mother did not showed neuropsychiatric manifestations. According to the clinical manifestations and genetic analysis, a pedigree of X-ALD was constructed. The patient was administrated with hydrocortisone 20 mg per day, mecobalamine, and compound vitamin B. During the fellow-up, the proband showed rapidly progressed neuropsychiatric manifestations including blurred vision, instability of gait, and incontinence within six months.
Case 2
A 31-year-old male was presented with adrenal crisis, exhibiting severe fatigue, anorexia, nausea and vomiting five years ago. The patient had significantly generalized skin pigmentation especially in the gingiva, areolae, and creases of the hands. Laboratory examination revealed severe hyponatremia (serum sodium 108 ~ 125 mmol/L, normal range 145–155 mmol/L), markedly decreased cortisol level (24.83-45.40-35.13 nmol/L) and increased ACTH level (1522-1071-1750 pg/mL) at 8am-4 pm-0am, respectively. The adrenal CT scan showed bilateral adrenal atrophy. At first, the patient was diagnosed as adrenal insufficiency, without exhibiting central or peripheral neuropsychiatric symptoms. After therapy with hydrocortisone (20 mg 8am,-10 mg 4 pm), symptoms of fatigues, vomiting were improved. The patient showed incorrigible severe hyponatremia, only corrected into normal by treated combination with fludrocortisone (0.05 mg per day). The serum sodium levels were maintained in normal range for five years. However, in recent eight months, the patient showed significantly aggravated walking instability, cognitive dysfunction, intellectual disabilities, emotional disturbance, memory loss and incontinence. Neurological physical examination revealed positive Barbinski’s sign and Hoffman’s sign, hyperactive tendon reflexes, and abnormal heel-to-knee-to-toe test. He spoke unclearly with dysarthria, while the vision and hearing were normal. Brain MRI revealed long T1 and long T2 signals in white matter of bilateral frontal lobes, and around the anterior of lateral ventricle, and corpus callosum (Fig. 1B). The patient was sickness and showed poor learning ability during childhood. Gene sequencing showed a heterozygous deletion mutation c.96_97delCT in ABCD1 gene (Fig. 1C and 1D), leading to the frameshift and premature transcription termination of amino acid p.Tyr33Profs*161. His immediate family members (parents and his sister) refused to perform genetic sequencing. His brother also showed sickness in childhood, and died at his eight-year-old. His maternal uncle was weak and fatigued in his youth, and showed disturbance of consciousness from 25-year-old, and died at 28-year old. None of the other family members exhibited neuropsychiatric symptoms and signs of X-ALD. The pedigree of X-ALD was suspected. During the fellow-up, the patient was treated with hydrocortisone and fludrocortisone, and serum sodium level was maintained in normal range. However, he showed rapidly progressed symptoms of incontinence and consciousness dysfunction.