3.1 Characterization of the lysine-malonylated proteins in S. aureus
S. aureus cells were collected during exponential phase and western blot analysis of whole cell lysates was carried out using pan-anti-acetylation, -2-hydroxyisobutyrylation, -succinylation, and -malonyl-lysine antibodies. As depicted in Fig. 1, lysine malonylation is widely spread in the proteome of S. aureus. For detecting whether the malonylated antibody has any cross-reactivity with other closely related lysine acylations (such as succinylation, glutarylation and acetylation), further research is needed. Global malonylome analysis of S. aureus proteome was performed using affinity enrichment followed by high-resolution LC-MS/MS (Fig. S1). Investigation of peptide distribution showed that the length of most peptides was in the range of 8–20 (Fig. S2), thus confirming adequate sample preparation.
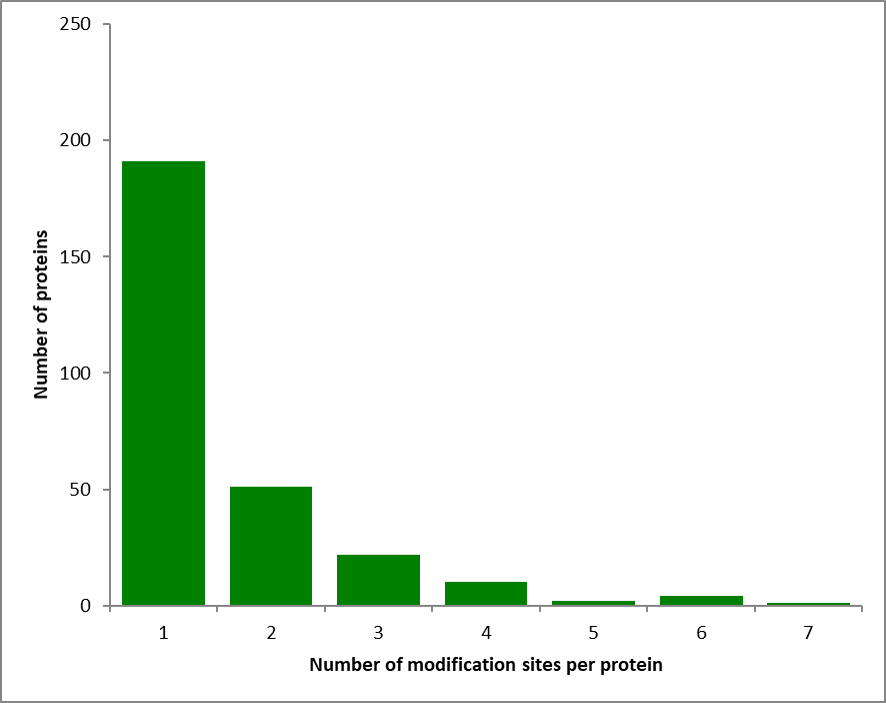
A total of 440 Kmal sites were identified in 281 proteins of S. aureus (Supplementary Table S1), which is fewer compared to the number of malonylated proteins recently identified in E. coli and B. amyloliquefaciens[14], but greater than in S. erythraea[15] and Toxoplasma gondii[26]. Among malonylated proteins of S. aureus, 191 proteins (67.9%) contained a single Kmal site, 51 proteins (18.1%) contained two Kmal sites, and the remaining had three or more Kmal sites (Fig. S3). Dihydrolipoyl dehydrogenase contained the largest number of malonylated sites (n = 7) withing a single protein. The second highly malonylated proteins contained six Kmal sites, including phosphoglycerate kinase (PGK) and alanine dehydrogenase (ALD), which are involved, respectively, in the second stage of glycolysis and oxidative deamination of alanine. Lysine malonylation was also found to happen on certain proteins of the 50S ribosomal family (L1, L9, L3, L6, L30), and five Kmal sites were also identified in heat shock protein 70 (Hsp70) in this study. Previous studies have shown that heat shock proteins are highly succinylated, containing up to 17 independent lysine residues and are crucial for host immune response regulation during infection by Plasmodium falciparum[27]. Dihydrolipoyl dehydrogenase discussed previously was the most intensively acetylated proteins with 15 Kmal sites in Saccharopolyspora erythraea.
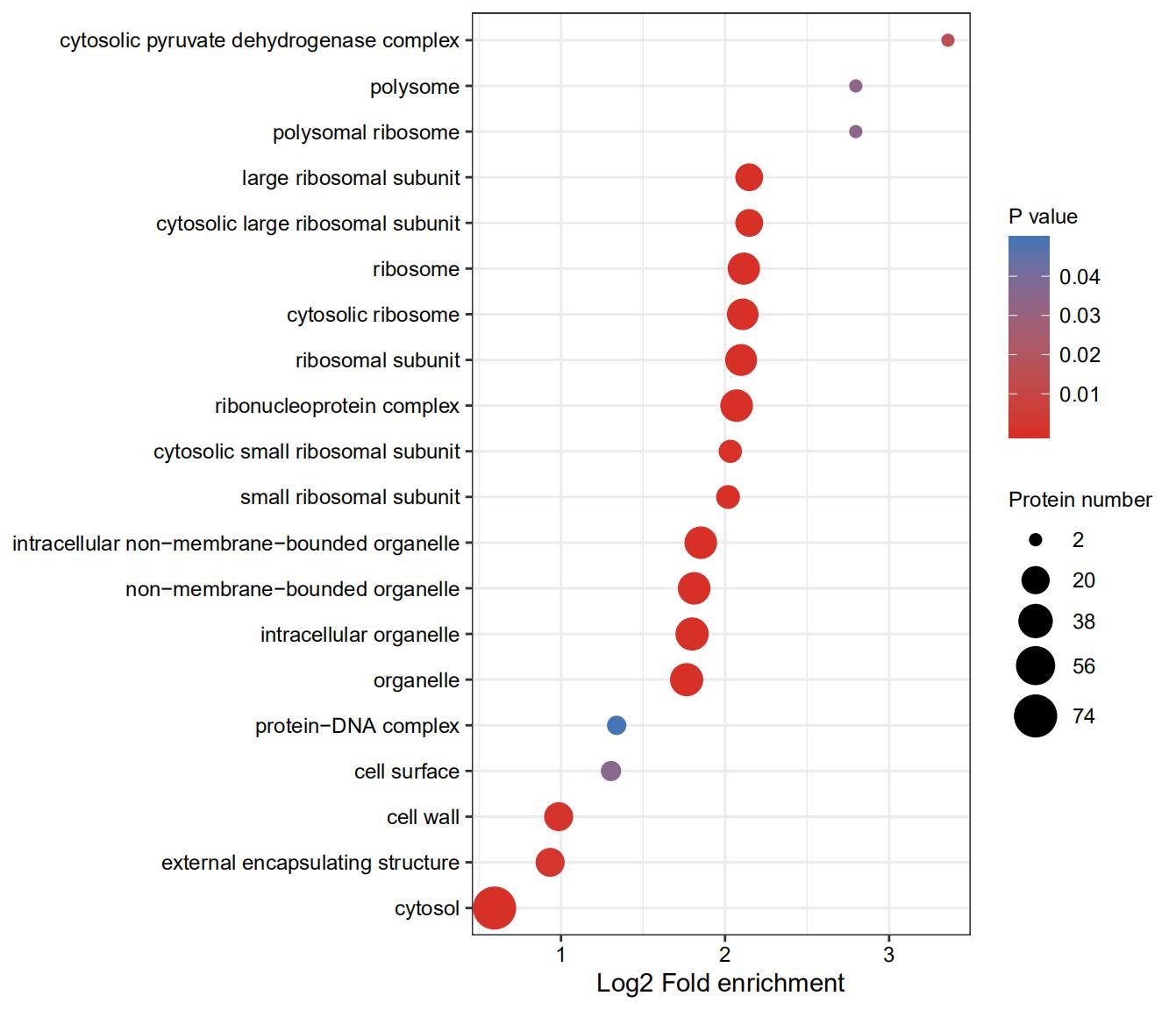
GO analysis revealed that malonylated proteins of S. aureus were shown to be preferred located in the cytosol (54.7%) (Supplementary Fig. S4), or as intracellular non-membrane-bounded organelle (11.3%), ribonucleoprotein complex (14.8%), cytosolic ribosome (9.6%), and ribonucleoprotein complex (9.6%). Several malonylated proteins have been identified in the nucleus, cytoplasm, mitochondria and chloroplast, thus indicating that a wide variety of biological processes can be potentially regulated by lysine malonylation.
3.2 Malonylated proteins in S. aureus are involved in central carbon metabolism
A growing body of evidence suggests that lysine malonylation plays a major role in metabolism regulation in bacteria. Malonylome analysis of mammalian cells revealed that SIRT5 regulates both cytosolic and mitochondrial proteins with glycolysis as the targeted pathway. KEGG pathway analysis showed that six categories were highly enriched (Fig. 2): ribosome, glycolysis/gluconeogenesis, pentose phosphate pathway (PPP), tricarboxylic acid cycle (TCA), valine, leucine, isoleucine degradation, and aminoacyl-tRNA biosynthesis. Malonylated proteins related to the ribosome pathway were significantly enriched, suggesting a potential involvement of lysine malonylation in protein synthesis. Enrichment of glycolysis/gluconeogenesis, pyruvate metabolism, and citric acid (TCA) cycle KEGG pathways were also observed in E. coli, S. erythraea, Fragaria vesca, and human cells, suggesting that malonylation may control activity or stability of enzymes involved in those pathways and thus impact regulation of energy metabolism.
Interestingly, nearly all enzymes involved in glycolysis were malonylated at one or more sites (Fig. 3), including critical enzymes 6-phosphofructokinase (PfkA), PGK, and pyruvate kinase (PYK); the latter was found to be malonylated at K66, K433, K434, K435, and K437. Fructose-bisphosphate aldolase plays a central role in glycolysis/gluconeogenesis and may serve as a potential target to fight pathogenic bacteria, and was also found to be malonylated in S. aureus. Moreover, other six malonylated proteins were found to belong to the pentose phosphate pathway. For instance, LpdA, a subunit of pyruvate dehydrogenase, contained 7 Kmal sites, being also highly malonylated in E. coli (15 Kaml sites). Additionally, three enzymes involved in the TCA cycle, including succinyl-CoA synthetase, fumarate hydratase, and malate dehydrogenase, were also lysine-malonylated in S. aureus. Other malonylated enzymes in S. aureus are involved in pyruvate metabolism, namely dihydrolipoyl dehydrogenase (PdhD), LpdA, fructose-bisphosphate aldolase (ALDO) (Fig. 3). Our findings were consistent with previous results in other prokaryotes for which malonylome analysis has been conducted[28]. Considering that the above-mentioned enzymes are mainly related to energy biosynthesis, malonylation is likely to trigger energy generation in bacteria.
Acyl-lysine modification can regulate protein-protein interaction. PPI analysis in the STRING database and PPI networks visualized in Cytoscape helped identify major biological processes affected by Kmal in S. aureus (Fig. 4). A number of highly associated subnetworks of Kmal proteins was revealed, which included glycolysis/gluconeogenesis and ribosome-related processes, which is consistent with KEGG pathway enrichment analysis.
3.3 Enrichment analysis of S. aureus lysine-malonylated proteins
Enrichment analysis was performed to determine functional categories for lysine-malonylated proteins in S. aureus. Most malonylated proteins in S. aureus were significantly related to the following categories: ligase activity, small molecule binding protein, and structural constituent of ribosome (Fig. 5A). Other categories, such as organic cyclic compound binding, heterocyclic compound binding, nucleic acid binding, structural constituent of ribosome, RNA binding, aminoacyl-tRNA synthetases, and elongation factors (FusA, TufA, Efp) were also significantly enriched, suggesting a role of protein malonylation in protein synthesis in S. aureus. Additionally, DnaK and Tig, two proteins involved in protein folding and export, respectively, were also malonylated in S. aureus. DnaK in Salmonella Typhimurium was also found to harbor two Kmal sites (K324, K555)[29]. Synthesized proteins that undergo further modification by chaperones need to find proper localization in the cell by chaperone-mediated transportation. This evidence suggests a potential role of lysine malonylation in controlling protein synthesis in S. aureus.
DNA gyrase subunit A (GyrA) in S. aureus was found to be malonylated with one modification site. The malonylated protein fructose-bisphosphate aldolase, which plays an essential role in glycolysis and gluconeogenesis pathways, has been considered a potential target for drug development against pathogenic bacteria[30], were malonylated containing 2 Kmal sites in K264 and K265. In addition to the regulation of cell metabolism and protein synthesis, lysine malonylation also modulates gene expression.
In this study, RNA polymerase subunits RpoA, RpoB, and RpoC were found to contain Kmal sites. RNA polymerase subunits were also found to be highly acetylated (15 sites in RpoB, 11 in RpoC, and 2 in RpoA) in E. coli[31]. Moreover, proteins involved in Rho-dependent termination were also lysine-malonylated in S. aureus, suggesting a potential role of lysine malonylation in controlling RNAP promoter specificity, strength, and activity. Additionally, primary free radical scavenging enzymes in S. aureus, such as superoxide dismutase, alkylhydroxide peroxidase, thioredoxin, and catalase also contained malonylated modifications.
To identify lysine-malonylated residues in protein active sites, protein domains and functional sites were annotated by InterProScan based on protein sequence alignment using the InterPro database. Among malonylated peptides in S. aureus, the following signatures and active site motifs were found (Supplementary Table S2): 30S ribosomal protein S3 site in KH domain; Thiol reductase thioredoxin site in thioredoxin; alanine dehydrogenase site in alanine dehydrogenase/PNT, N-terminal domain; pyruvate kinase site in pyruvate kinase alpha/beta domain PYK; pyruvate carboxylase site, biotin carboxyl carrier protein of acetyl-CoA carboxylase site in biotin-requiring enzyme; catabolite control protein site in transcriptional regulator CcpA; pyruvate oxidase site in thiamine diphosphate-dependent CidC; dihydrolipoyl dehydrogenase site in pyridine nucleotide-disulphide oxidoreductase LpdA. Several malonylated peptides were also found as signatures in PnpA (an RNA binding domain profile), GlyS (Anticodon binding domain profile), ALD (alanine dehydrogenase/PNT profile), RplA (ribosomal protein L1p/L10e family) and TufA (elongation factor Tu domain) (Fig. 5B). Those findings pinpoint the preferred location of malonyl groups in S. aureus proteins and suggest possible specialized functions of malonylation.
3.4 Pattern analysis of malonylated peptides
Structural analysis of malonylated peptides in S. aureus enabled identification of surface accessibility of Kmal sites. The average surface accessibility of malonylated lysines was significantly higher (p < 0.005) than non-malonylated lysines (Fig. 6A), which also indicates that Kmal sites are preferably located on the surface of proteins. Certain binding proteins may increase hydrophobicity of large complexes, thereby interfering with normal assembly in an aqueous environment. Lysine malonylation may thus balance hydrophobicity by enhancing accessibility to protein surface. Interestingly, 32.96% of Kmal sites were located in secondary structure regions; of them, 26.55% were located in α-helices and 6.41% in β-sheets regions. The remaining 67.02% of malonylation sites in S. aureus proteins were located in coil unstructured regions (Fig. 6A), suggesting that malonylation is more likely to occur in disordered rather than ordered regions in proteins.
Several studies on prokaryotes have proved preferences for amino-acid residues at particular positions surrounding the acetylated lysine and succinylated lysine[32]. Malonyl-proteins regulated by the same type of enzymes often exhibit similar sequences. Therefore, amino acids around the malonylated lysine from − 10 to + 10 were mapped to test if there are specifific amino acids adjacent to malonylated lysines. As shown in Fig. 6C, the frequency of valine (V) in position -1 and alanine (A) at +2 and +4 position was high, which is in agreement with heat map analysis (Fig. 6B). Alanine (A) was overly represented around the malonylated lysine in S. aureus, a pattern that is similar to Kmal found in E. coli[13].
3.5 Conserved lysine-malonylated proteins in S. aureus and E. coli
Lysine malonylome of S. aureus was compared to previously reported E. coli malonylomes. Functional classification of S. aureus lysine-malonylated proteins is very similar to that of E. coli[13]. Interestingly, 31 malonylated sites in S. aureus were considered homologous with E. coli malonylome (Fig. 7; Supplementary Table S3). Most of acetylated proteins involved in protein synthesis and energy metabolism in S. aureus were considered orthologous to those of E. coli. A small subset of lysine-malonylated proteins was commonly found in these two bacterial malonylomes: DNA-binding protein, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, NADP-dependent phosphogluconate dehydrogenase, ATP synthase, 50S ribosomal protein L1, 50S ribosomal protein L3, glyceraldehyde-3-phosphate dehydrogenase, 30S ribosomal protein S3, dihydrolipoyl dehydrogenase, pyruvate kinase, and formate C-acetyltransferase.
{kind=link}
{kind=link}
{kind=link}
{kind=link}