Materials and Methods
All chemicals used throughout this investigation were purchased from Sigma Aldrich, Fluka and Merck, and were used as received. Progress of reaction and purity of products were monitored by thin layer chromatography (TLC) carried out on packed on silica gel Al (aluminum) sheets (60 F254). Plates were visualized under UV light, where appropriate. Catalyst was characterized by means of a scanning electron microscope (SEM), with accelerating voltage 0.2–30 kV of 50/60 Hz power by (Carl Zeiss Company). Infrared (FTIR) spectra were recorded as KBr pellets in the range of 650-4000 cm–1 with a Shimadzu Corporation. 1H NMR spectra of prepared compounds were recorded with the aid of a 500 MHz spectrometer (AVANCE AV). Chemical shifts are reported in ppm using tetramethylsilane (TMS) as an internal standard and CDCl3 as solvent. (1H NMR spectra of prepared compounds are displayed in Fig. 5S-Supplimentary).
1H NMR spectra of prepared compounds are displayed in the supplementary material.
Catalyst preparation
Catalyst was prepared according to a published procedure which involved stirring a mixture of conc. sulfuric acid and silica gel in dichloromethane for 4 h; this has afforded a white amorphous powder, which was used as a solid support medium without further purification (Riego, Sedin, Zaldívar, Marziano, & Tortato, 1996).
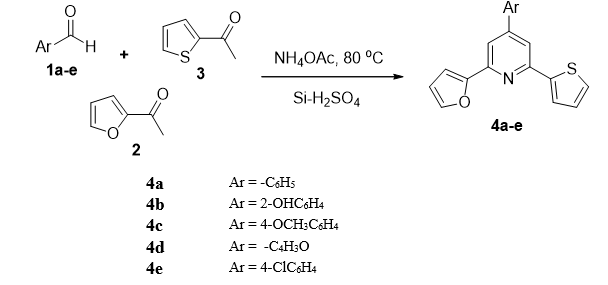
General procedure for preparation of 4a–e
Compounds 4a–e were prepared by the following procedure: Equimolar amounts of arylaldehydes (1a–e), 2-acetylfuran (2), 2-acetylthiophene (3), NH4OAc (1.5 mole %), and an appropriate amount of silica catalyst were mixed in a round bottom flask and refluxed at 80 oC for 1 h (Table 1). Progress of reaction was monitored by TLC using (10 %) a mixture of n-hexane and ethyl acetate as a solvent system. Upon completion of reaction, the mixture was allowed to cool to room temperature, dissolved in methanol, and filtered. The filtrate was poured onto crushed ice to afford the final products (4a–e). Using the same general procedure, the following compounds were prepared:
2-(Furan-2-yl)-4-phenyl-6-(thiophen-2-yl) pyridine (4a)
Prepared as light brown sticky solid, yield 60 %; mp 113 oC; Rf = 0.72 cm–1; IR (KBr) cm–1: 1559 (C=C), 1493 (C=N-C), 1235 (C-O-C), 1161 (C-S-C); 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.85 (1H, d, J = 2.1 Hz, H-3 pyridyl), 7.82(1H, d, J = 2.1 Hz, H-5 pyridyl), 7.62–7.78 (3H, m, Ar-H furfuryl), 7.40–7.32 (3H, m, Ar-H thienyl), 6.55–6.52 (5H, m, Ph),; MS m/z (ESI-MS) 304 (M+) (90 %).
2-(2-(Furan-2-yl)-6-(thiophen-2-yl) pyridin-4-yl) phenol (4b)
Dark brown, gummy solid; yield 65 %; mp 112° C; Rf = 0.846 cm–1; IR (KBr) cm–1: 1559 (C=C), 1490 (C=N-C), 1235 (C-O-C), 1157 (C-S-C); 1H-NMR (500 MHz, CDCl3) δ (ppm):
7.74 (1H, d, J = 2.2 Hz, H-3 pyridyl) 7.63 (1H, d, J = 2.3 Hz, H-5 pyridyl), 7.61–7.52 (3H, m, Ar-H furfuryl), 7.35–7.31 (3H, m, Ar-H thienyl) 6.99 (1H, dd, J = 8.2, 2.1 Hz, Ph H-3), 6.57 (1H, ddd, J = 8.3, 8.3, 2.1Hz, Ph H-5), 6.55 (1H, dd, J = 8.2, 8.1 Hz, Ph H-4), 6.53 (1H, dd, J = 8.2, 2.1 Hz, Ph H-6). ; MS m/z (ESI-MS) 320 (M+) (90 %).
2-(Furan-2-yl)-4-(4-methoxyphenyl)-6-(thiophen-2-yl) pyridine (4c)
Light brown, sticky solid; yield 63 %; M.P. 110° C; Rf = 0.57 cm–1; IR (KBr) cm–1: 1559 (C=C), 1510 (C=N-C), 1243 (C-O-C), 1172 (C-S-C); 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.85 (1H, d, J = 2.2 Hz, H-3 pyridyl), 7.83 (1H, d, J = 2.2 Hz, H-5 pyridyl), 7.62–7.59 (3H, m, Ar-H furfuryl), 7.33-7.28 (3H, m, Ar-H thienyl), 7.15 (2H, d, J = 8.2 Hz, H-2 and H-6), 6.92 (2H, d, J = 8.2 Hz, H-3 and H-5), 3.86(3H, s, OCH3); MS m/z (ESI-MS) 334 (M+) (100 %).
2,4-Di(furan-2-yl)-6-(thiophen-2-yl) pyridine (4d)
Dark brown, sticky solid; yield 60 %; M.P. 145° C; Rf = 0.57 cm–1; IR (KBr) cm–1: 1559 (C=C), 1507 (C=N-C), 1237 (C-O-C), 1156 (C-S-C); 1H-NMR (500 MHz, CDCl3) δ (ppm): 7.62 (1H, d, J = 2.2 Hz, H-3 pyridyl), 7.60 (1H, d, J = 2.2 Hz, H-5 pyridyl), 7.79–7.73 (3H, m, Ar-H furfuryl), 7.32–7.29 (3H, m, Ar-H thienyl), 6.96–6.70 (3H, m, Ar-H furfuryl); MS m/z (ESI-MS) 294 (M+) (99 %).
4-(4-Chlorophenyl)-2-(furan-2-yl)-6-(thiophen-2-yl) pyridine (4e)
Light brown, granular solid; yield 55 %; M.P. 120° C; Rf = 0.65 cm–1; IR (KBr) cm–1: 1559 (C=C), 1490 (C=N-C), 1247 (C-O-C), 1163 (C-S-C); 1H-NMR (500 MHz, CDCl3) δ (ppm):
7.81 (1H, d, J = 2.2 Hz, H-3 pyridyl), 7.78 (1H, d, J = 2.2 Hz, H-5 pyridyl), 7.64–7.57 (3H, m, Ar-H furfuryl), 7.42-7.35 (3H, m, Ar-H thienyl), 6.59 (2H, d, J = 8.2 Hz, H-3 and H-5), 6.38(2H, d, J = 8.2 Hz, H-2 and H-6); MS m/z (ESI-MS) 338 (M+) (25 %).
Biological Activities
In Vitro Cytotoxicity Assay
Cytotoxicity of the newly synthesized 2,4,6-triarylpyridine derivatives has been performed in 96-well microtiter plates using the 3-[4, 5-dimethylthiazole-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) colorimetric assay. In this assay, mouse fibroblast (3T3) cells were cultured in Dulbecco’s Modified Eagle Medium, and mixed with 10 % of fetal bovine serum (FBS), 100 IU/mL of penicillin, and 100 1g/mL of streptomycin in 75 cm2 flask. Flasks were then kept in an incubator with 5% CO2 at 37 °C. Cultured cells with a concentration of 5, 9, and 104 cells/mL were introduced (100 µL/well) into 96 well plates. After an incubation period of 12 h, 200 µL of fresh medium was added with different concentrations of compounds 4a–e (1 M). After 2 days, 200 µL of MTT (0.5 mg/mL) was added to each well, and kept incubated for 4 h. This was followed by addition of 100 µL of DMSO to each well. The degree of reduction of MTT to form a zone within cells was measured at 540 nm using a microplate reader. Cytotoxicity profile was calculated as the concentration causing 50% growth inhibition (IC50) of 3T3 cells (Saleem et al., 2016). The percent inhibition was calculated by using the following formula:

Antibacterial Screening
We evaluated the antibacterial activity of synthesized compounds 4a–e using the disc diffusion method. Briefly, test compounds (10 mg/mL) were applied on the pasteurized filter paper discs, dried overnight at room temperature, and stored; the negative control discs were prepared using the same procedure. In a standard microbiological working environment, bacterial cultures were grown overnight at 37 oC in nutrient broth medium and spread onto solidified nutrient agar medium in petri dishes. Then control and test discs were applied onto the solidified medium surface and incubated at 37 oC for 12–15 h. Results were calculated by evaluating the zone of inhibition in millimeters for each compound and compared with sparaxin, used as a standard (Pervez et al., 2008).
Urease Inhibition
Investigation of the urease inhibition activity of compounds 4a–e was carried according to the procedure outlined by Khan (2017) with slight modifications. Briefly, a total 200 µL (0.5 mM) reaction mixture containing a 25-µL urease enzyme solution was incubated for 15 min at 30 oC. Then, 55 µL of buffer and 100 mM of substrate urea were incubated at the same conditions. After that, 0.5% w/v sodium hydroxide, 0.1 % sodium hypochlorite (70 µL), 1 % w/v phenol, and 0.005 % w/v sodium nitroprusside (45 µL) reagent were added and incubated at 30 oC for 50 min. To conclude the urease inhibition activity, ammonia production was measured by the indophenol method. Optical density (OD) measurements were conducted at 630 nm with the aid of an ELISA plate reader (KHAN, 2017). In this assay, thiourea was used as a standard and the percentage of inhibition was calculated using the following formula:


{kind=link}