Gene amplification of FAM3C and MET is tightly linked in several human carcinomas and correlates with increased gene expression and poor prognosis
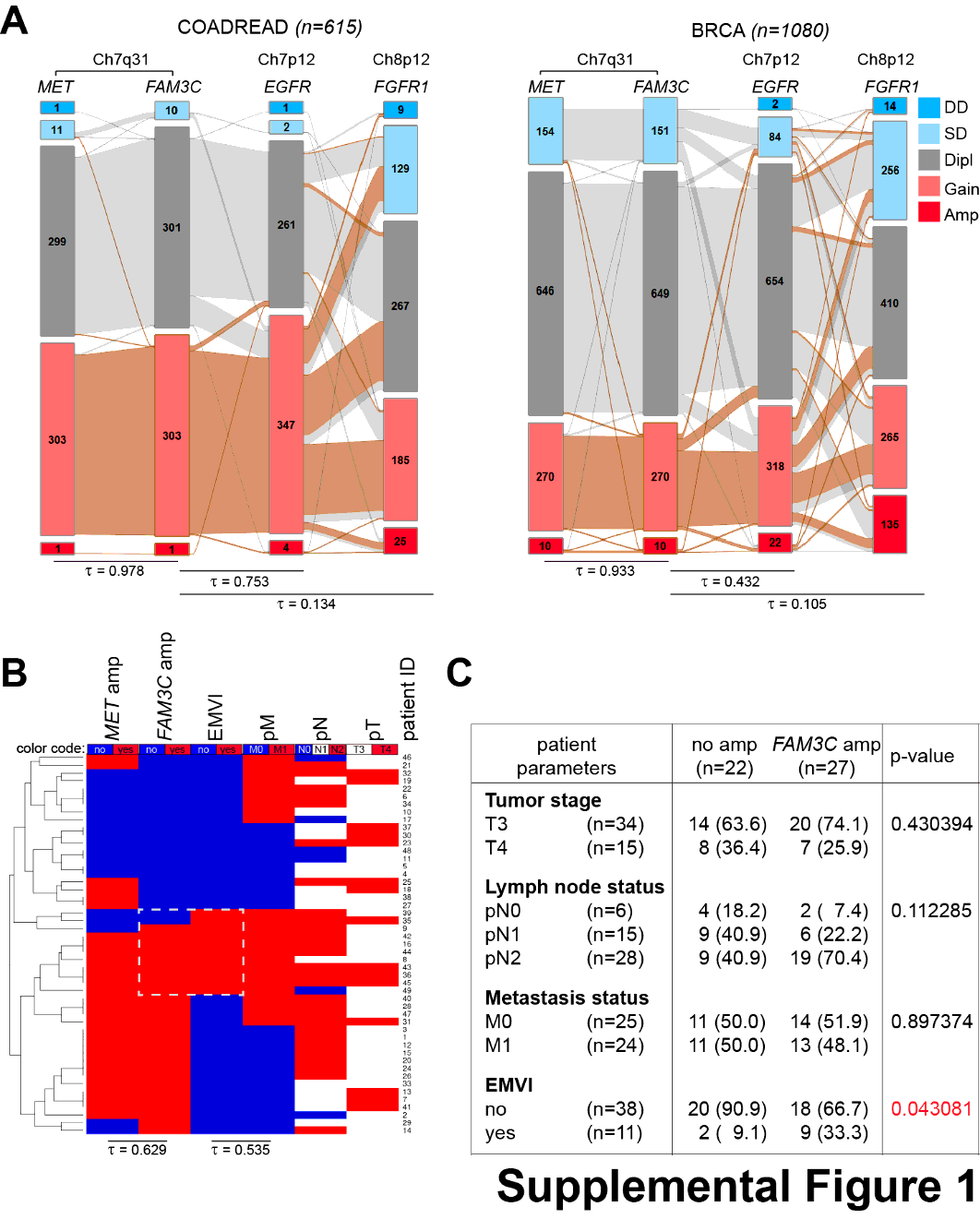
To determine the frequency of CN amplification of the FAM3C and MET genes, we investigated a variety of datasets of different cancer entities from the TCGA database. Connections in gene function and physical location of the two genes were uncoupled by analysis of two additional RTKs with dedicated driver functions in the progression of many malignancies: Epidermal growth factor receptor (EGFR), located on the p arm of the same chromosome and fibroblast growth factor receptor 1 (FGFR1) located on chromosome 8. Of 501 lung squamous cell carcinoma (LUSC) cases, 8 (1.9%) indicated MET, out of these 6 (1.4%) also FAM3C CN amplifications (Fig.1A). Of 516 lung adenocarcinoma (LUAD) cases, 18 (3.5%) had MET and 9 (1.7%) FAM3C CN amplification, 8 deriving from the MET amplification group (Fig.1D). 1480 hepatocellular carcinoma (LIHC) cases had in 11 (0.7%) and 6 (0.4%) cases with amplification for the MET and FAM3C genes, respectively, in 5 cases with a shared amplification for both, 615 colorectal adenocarcinoma (COADREAD) cases showed CN amplification in 1 case (0.2%) shared for both MET and FAM3C and 1080 breast cancer (BRCA) cases had CN amplification in 10 cases (0.9%) for both MET and FAM3C, 7 sharing amplification for both (Supplemental Fig.S1A). Correlation analyses showed that the FAM3C gene CNs were tightly correlated with CNs of MET but not with the distant EGFR or unlinked FGFR1 genes in all analyzed data cohorts for LUSC (Fig.1A), LUAD (Fig.1.D), LIHC, COADREAD, and BRCA (Supplemental Fig.S1A), indicating that co-amplification might be a consequence of chromosomal proximity.
To test if genomic amplification influenced gene expression, MET and FAM3C gene CNs were then compared to mRNA levels and relapse-free survival of LUSC and LUAD patients from the TCGA database. The analysis showed that CNs of both genes significantly correlated with gene expression levels, patients with strong MET and/or FAM3C amplification showing the highest expression of these genes (Fig.1B and E). Patients with MET and/or FAM3C amplification also had a significantly worse survival compared to the pooled cohort of patients with deletions, normal, or slight gain in the CN of the two gene loci, albeit low case number and early loss on patient follow-up prevented a proper analysis on the survival of LUSC FAM3C amplified patients (Fig.1C and F). These data indicate that the FAM3C and MET genes are frequently co-amplified in human cancers contributing to increased gene expression and poor survival.
We then tested the linkage of the two genes on genomic DNA isolated from formalin fixed paraffin-embedded tumors of 49 advanced-stage colorectal carcinoma patients by qPCR. Over 72% (24/33) of the FAM3C- and/or MET-amplified tumors showed a co-amplification of the two genes, further supporting the tight linkage of these loci (Supplemental Fig.S1B). In addition, cluster and correlation analysis of FAM3C amplification with available clinicopathological parameters elucidated a significant enrichment of FAM3C amplification in patients with extramural venous invasion (EMVI) (Supplemental Fig.S1B, C). EMVI, the spreading of cancer cells into the nearby blood vessels, is an invasive characteristic connected to worse prognosis. Although ILEI has not been linked so far to EMVI, our finding is in accordance with the described function of ILEI in inducing EMT and invasion and reflects that FAM3C amplification might affect gene function resulting in a clinically worse outcome. So, these results support the database analysis showing frequent co-amplification of MET and FAM3C and the likelihood of poor survival rates in patients with increased CNs of these genes.
Increased CNs of FAM3C and MET are tightly linked and frequently present in multiple human cancer cell lines
To evaluate if our findings on FAM3C-MET co-amplification in human primary tumors can be recapitulated in cultured human cancer cell lines, FAM3C and MET gene CNs were determined in a panel of 200 human cancer cell lines of diverse tissue origins by microarrays and analysed using the CNAT 4.0 analysis algotrythms in the GTC analysis (Fig.2A). Increased CN, using a cut-off of CN 3 or higher, of both genes was present in cell lines of all tumor entities at a frequency of 47% on average. The frequency varied in the different cancer types, breast and lung cancer had the lowest (25% and 30%) and melanomas the highest (76%) (Fig.2A, left panel). Importantly, over 90% of the cell lines with an increased CN for at least one of the genes showed an increase for both loci (Fig.2A, right panel), confirming that the amplification event of the two genes is tightly coupled and indicating that cancer cell lines representatively illustrate in vitro the FAM3C-MET co-amplification characteristics of primary tumors.
Of the 85 cell lines with increased CN for both FAM3C and MET, we selected five for detailed investigation (Fig.2B). These were a pair of gastrointestinal cancer cell lines (MKN45 and OE33), a pair of lung adenocarcinoma cancer cell lines (NCI-H1993 and NCI-H441), and a breast cancer cell line (SKBR3). One cell line in each of the gastrointestinal and lung adenocarcinoma pairs had previously been described as sensitive and the other as resistant to the c-MET inhibitor PHA665752 (19). Both these pairs of cell lines expressed high levels of ILEI and c-MET (Fig.2C) as compared to control samples from non-metastatic MCF7 and the metastatic MDA-MB-231 human breast cancer cell line lacking FAM3C and MET amplifications but with upregulated ILEI expression as a characteristics of metastatic capacity (4). These cell lines with high expression levels also secreted ILEI into the CM during culture. In SKBR3 cells, c-MET expression is absent despite locus amplification due to epigenetic silencing (5). Accordingly, this cell line did not express c-MET and interestingly, though not silenced, ILEI expression was also only moderate and secretion almost absent despite increased gene CN (Fig.2C).
Stable ILEI knock-down does not influence proliferation capacity and sensitivity towards c-MET-inhibitor induced proliferation arrest
Since there are no specific pharmacological inhibitors for ILEI available, we mimicked ILEI inhibition by RNA interference-mediated (RNAi) stable knock-down (KD) of the protein expression. Both intracellular and secreted ILEI protein levels showed an apparent reduction by two independent shRNAs in all five cell lines, which was most evident in the levels of the functionally relevant secreted form in the CM (Fig.3A). For cMET blockade, we used crizotinib, a small-molecule tyrosine kinase inhibitor that efficiently inhibits c-MET, anaplastic lymphoma kinase 5 (ALK5) and ROS1 and is approved by the FDA for treatment of ALK-rearranged NSCLC (4). As none of the selected cell lines including the two lung adenocarcinoma lines NCI-H1993 and NCI-H441 harbored an ALK rearrangement, inhibitor effects were expected to occur primarily due to c-MET inhibition. Since SKBR3 cells did not express c-MET despite of a MET and FAM3C amplification, they served as a control to monitor the potential influence of ALK5 and ROS1 targeting effects of crizotinib.
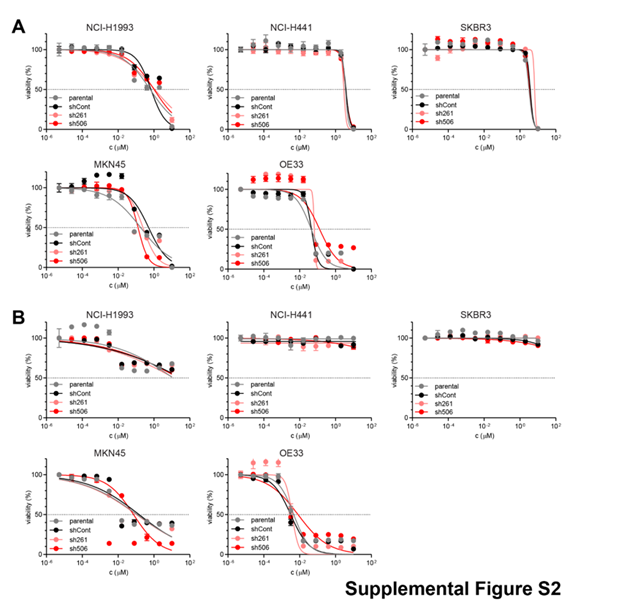
First, we analyzed the dose-dependency of crizotinib on the proliferation capacity of the five selected cancer cell lines. The OE33 cell line that had previously shown resistance to another small molecule c-MET inhibitor displayed sensitivity to increasing concentrations of crizotinib in proliferation capacity (Fig. 3B), comparable to the sensitive cell lines NCI-H1993 and MKN45. NCI-H441 and SKBR3 tolerated high doses of the drug without remarkable drop in their proliferation rate or viability (Fig.3B), confirming their described resistance towards MET inhibitors (19). ILEI KD did not influence the proliferation behavior of the selected five cell lines (Fig.3C). Furthermore, ILEI KD also did not influence the sensitivity of these cells towards crizotinib-induced growth arrest (Fig.3C), indicating that ILEI does not affect proliferation and does not influence c-MET-dependent regulation of proliferation. To address any concerns of the polypharmacological action of crizotinib, which inhibits other targets such as ALK5, we also investigated the action of two additional c-MET inhibitors: PHA665752 and savolitinib. The results showed similar effects on cell viability as crizotinib (Supplemental Fig.S2).
ILEI KD impairs both c-MET-independent and c-MET-dependent invasion of cancer cells with FAM3C and MET CN gains
Next, we investigated invasiveness and its sensitivity to c-MET and ILEI signaling inhibition in the five selected cancer cell lines. First, we tested c-MET-independent invasive capacity in an in vitro trans-well invasion assay by using NIH3T3 CM as chemoattractant, as murine HGF produced by these cells does not cross-activate the human c-MET receptor (20, 21). ILEI KD strongly impaired the invasiveness of all five cancer cell lines, whereas invasion capacity was not influenced by crizotinib treatment (Fig.4A). This supported the view that ILEI signaling induces invasiveness. To test the influence of ILEI on c-MET induced invasion, the same assay was performed this time using human HGF as chemoattractant (Fig. 4B). As expected, crizotinib efficiently inhibited HGF-induced invasion in the four c-MET-expressing cell lines. Importantly, ILEI KD also significantly impaired HGF-induced invasiveness in all c-MET-expressing cells, indicating that ILEI might be a contributing factor in c-MET-driven cellular invasion. ILEI KD derivatives of NCI-H441 and OE33, even showed an increased sensitivity towards crizotinib with invasion almost completely eliminated, suggesting that ILEI depletion might have an additive inhibitory effect to crizotinib in these cells.
In summary, stable ILEI KD efficiently reduced both c-MET-dependent and c-MET-independent invasion in all tested cells.
While ILEI does not influence c-MET signaling activity, c-MET acts on ILEI signaling activity by regulating ILEI secretion
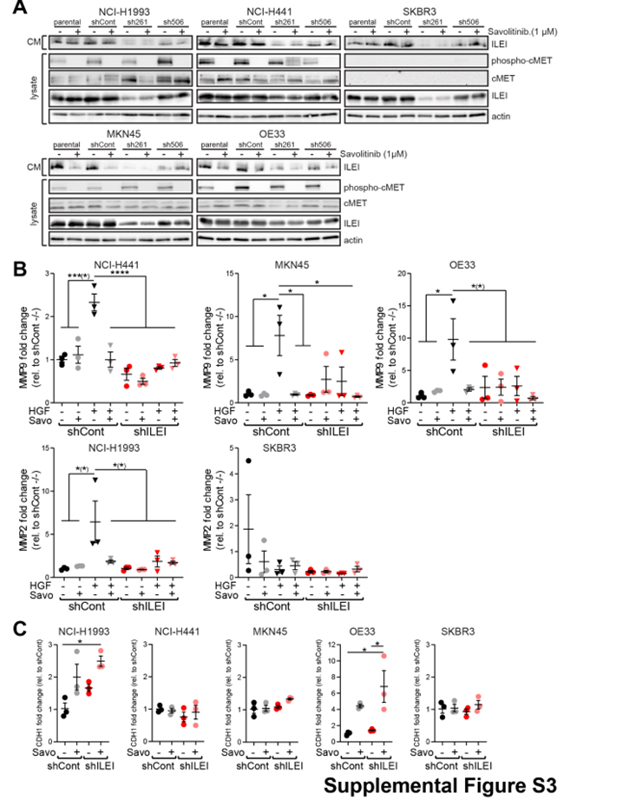
As c-MET and ILEI interact during c-MET-dependent invasion, we tested the possibility of the two signaling pathways being linked by investigating the effect of crizotinib and ILEI KD on c-MET signaling. Crizotinib efficiently inhibited c-MET autophosphorylation in all four c-MET-expressing cell lines (Fig. 5A). Activation of Erk, an important downstream effector of c-MET, was also significantly reduced upon drug treatment in these cells, whereas it remained unaltered in SKBR3 cells, which do not express c-MET (Fig. 5A). Knock-down of ILEI did not have an influence on the expression and activation levels of c-MET and Erk (Fig.5A). Similarly, c-MET inhibition did not influence ILEI expression levels in the tested cell lines (Fig. 5A), indicating that c-MET and ILEI expression is not cross-regulated. Importantly, however, crizotinib decreased the secretion of ILEI in all c-Met expressing cell lines, but not in SKBR3 cells (Fig. 5A). Similar results were also found with the c-MET specific inhibitor savolitinib (Supplemental Fig.S3A). This suggests that c-MET may positively regulate ILEI secretion.
Elevated expression and secretion of MMPs upon c-Met activation depends on ILEI and the two pathways cooperate in E-cadherin repression
To uncover potential mechanisms by which c-MET and ILEI might cooperate to increase invasiveness, we next investigated markers of invasion. MMPs remodel the extracellular matrix (ECM) and are activated during invasion to ease the movement of cancer cells (22). So, we explored the mRNA expression levels of two prominent MMPs, MMP-2 and 9 in response to HGF in the five cell lines. Of note, none of the five cells showed expression of both of these MMPs; NCI-H441, MKN45 and OE33 expressed only MMP-9, whereas NCI-H1993 and SKBR3 cells only MMP-2. Firstly, ILEI KD lead to decreased expression of MMP-9 or MMP-2 in all five cell lines, confirming its role in invasion and MMP expression (Fig. 5B) and (17). Secondly, in NCI-H441, MKN45, and OE33 cells HGF induced expression of MMP-9 mRNA and this expression was inhibited by crizotinib supporting the view that c-MET-dependent invasion activates MMP-9 (Fig. 5B) and (23, 24). Even more importantly, however, in ILEI KD derivatives of these cells HGF treatment was not able to elevate MMP-9 mRNA expression (Fig. 5B). This result suggests that c-MET-mediated increased expression of MMP-9 mRNA during invasion is dependent on ILEI. A similar pattern was seen in NCI-H1993 cells for MMP-2 mRNA expression, but not in SKBR3 cells, where c-MET is not expressed and hence, HGF did not increase MMP-2 mRNA levels (Fig.5B). As MMPs act on ECM, their activity is dependent upon secretion, so we analyzed the effect of c-MET and ILEI signaling inhibition on MMP secretion. For easier detection of inhibitory effects, high baseline secretion was ensured by HGF trigger. The secretion of MMP-9 from NCI-H441 cells decreased slightly with crizotinib treatment or ILEI KD, and the combination of the two lead to a significant reduction (Figs.5C, 5D). In NCI-H1993 cells ILEI KD was sufficient for remarkable reduction of MMP-2 secretion (Figs. 5C, 5D). The specificity of these results to c-MET inhibition was also tested with savolitinib with similar results (Supplemental Fig.S3B). Overall, these results suggest that both c-MET and ILEI contribute to efficient secretion of MMPs in a cooperative and partially complimentary manner, c-Met most probably acting indirectly, via regulating ILEI secretion.
Another important marker of invasion and EMT status is the loss or reduction of the cell adhesion molecule E-cadherin (25). Therefore, we checked potential changes in the levels of E-cadherin mRNA (CDH1) and protein upon crizotinib treatment and ILEI KD in each of the cell lines used in this study, and found high variance according to the cell type (Fig. 5E, F). On the one hand, in the crizotinib-resistant NCI-H441 and SKBR3 cells no significant differences in CDH1 mRNA expression levels were observed (Fig. 5E). On the other hand, NCI-H1993, MKN45 and OE33 cell lines showed a significant c-MET and ILEI mediated regulation of CDH1 transcription. All three cell lines showed increase in CDH1 mRNA levels in ILEI KD derived cells or with crizotinib, and combination of these conditions resulted in more stable or superior effects. Cells tested with savolitinib showed similar results (Supplemental Fig.S3C). E-cadherin protein levels showed a similar trend of differences as of transcription (Fig. 5F). This once again suggests a cooperation between c-MET and ILEI in the regulation of E-cadherin transcription, however, these data also point out that cancer cells show very different sensitivity towards this regulation.
Combined ILEI KD and crizotinib treatment significantly reduced the outgrowth of NCI-H441 and NCI-H1993 tumor xenografts
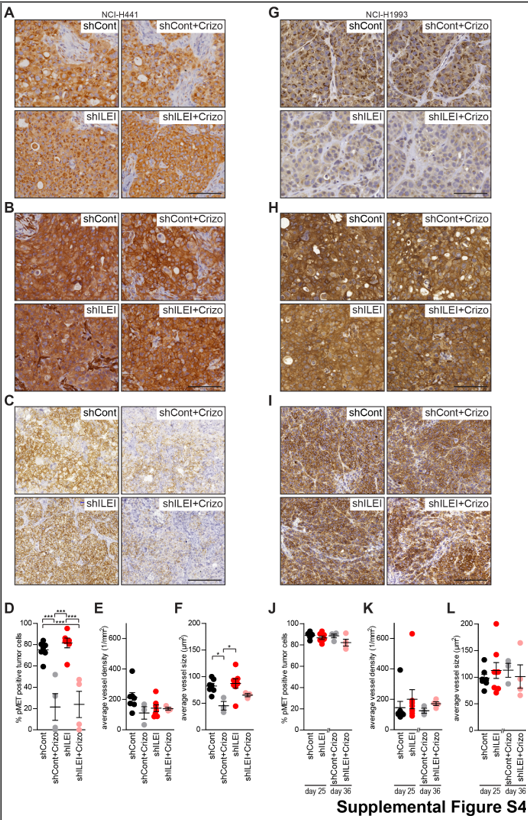
To assess the in vivo relevance of the above findings, we next investigated the growth of tumor xenografts induced by NCI-H441 and NCI-H1993 cells and their ILEI KD derivatives in the presence and absence of crizotinib treatment in a mouse model. The original rationale was to compare the effect of ILEI KD on the growth capacity of crizotinib-sensitive vs crizotinib resistant tumors. Because we were expecting a very low response of the NCI-H441 cell line towards crizotinib we did not plan a withdrawal step for that cell line, while the NCI-H1993 cell induced xenografts were evaluated for crizotinib withdrawal for an additional 11 days. However, in accordance with some earlier findings (26), we observed that inhibition of c-MET with crizotinib reduced the growth and tumor mass of both of the xenografts (Figs. 6A, 6B, 6J, 6K), indicating that crizotinib was able to counteract the resistance of NCI-H441 cells most probably via non-cell intrinsic mechanisms not addressed here. Importantly for this study, ILEI KD also slowed the growth of both xenografts. Tumor growth was most efficiently reduced when ILEI KD was combined with crizotinib (Figs. 6A, 6J). The tumor mass was significantly decreased in cells with ILEI KD compared to those with ILEI expression and was lowest in cells with ILEI KD and crizotinib combined (Figs. 6B, 6K). Immunohistochemistry for ILEI (Supplemental Fig. S4A,G), c-MET (Supplemental Fig. S4B,H), and phospho-cMET (Supplemental Fig. S4CI) confirmed significant ILEI KD, unaltered c-MET expression upon ILEI KD and inhibitor treatment, as well as efficient inhibition of c-MET activation by crizotinib, respectively. The latter was also quantified as percentage of phospho-c-MET positive tumor cells (Supplemental Fig. S4D,J). It is notable that decrease in c-MET activation was no more evident in the NCI-H1993 derived tumors due to crizotinib withdrawal over the last 11 days of the experiment (Supplemental Fig. S4J).
To investigate the reasons for smaller tumors resulting from crizotinib and ILEI KD we investigated the proliferation and apoptosis of the tumor cells by quantifying the percentage of Ki67 and activated Caspase3 positive cells on tissue sections, respectively. In NCI-H441 xenografts, crizotinib treatment significantly reduced the proliferation of cells, but ILEI KD alone did not influence this parameter (Fig. 6C). (26). So, this result supported the cell-based assays and showed that the cells were behaving in a similar manner in vivo to the in vitro analysis. In NCI-H1993 xenografts, the graph shows the recovery of the tumor cells after crizotinib treatment was halted on day 25 and observed for an additional 11 days. This indicated that after a period of drug withdrawal, proliferation was apparently no longer affected by the previous crizotinib treatment (Fig. 6L). Apoptosis was also decreased in NCI-H441 xenografts treated with crizotinib and in ILEI KD derived cells (Fig. 6D). In NCI-H1993 xenografts that had a withdrawal period from the crizotinib treatment phase there was no apparent significant difference in apoptosis with crizotinib or ILEI KD (Fig. 6M). So, the smaller tumors are likely to be due to the decreased proliferation upon crizotinib treatment rather than an increase in apoptotic cell death. A higher level of apoptosis in larger tumors without c-MET inhibition or ILEI KD may be indicative of the rapid turnover of cells in these rapidly proliferating tumors that was also manifested in a highly ulcerated appearance.
We also addressed if c-MET and ILEI had a consequence on tumor vascularization by determining blood vessel density and size on CD31 immunostained tumor sections. The vessel density remained constant between tumors (Supplemental Fig. S4E,K), and though NCI-H441 tumors showed decreased vessel size upon crizotinib treatment, it was less evident in the tumors with ILEI KD and not evident in any of the NCI-H1993 tumors (Supplemental Fig. S4F,L). These data indicate that decreased tumor size upon combined ILEI KD and c-MET inhibition is not primarily due to a switch in vascularization capabilities.
Combined ILEI KD and crizotinib treatment decreased MMP expression in NCI-H441 and NCI-H1993 tumor xenografts
To investigate whether the relationship between MMPs, c-MET, and ILEI seen in the cell lines was also evident in vivo, the expression of MMP-9 and MMP-2 from the tumors was investigated. In line with the results from gelatin zymography, the expression of MMP-9 protein in NCI-H441 tumors decreased slightly with crizotinib inhibition and even more when the cells also had ILEI KD, while MMP-2 expression, that became detectable only at in vivo conditions, was significantly decreased in ILEI KD tumors (Figs. 6E, 6F). At mRNA level, there was a significant decrease of MMP-9 in tumors from ILEI KD cells (Fig. 6G) and quantification of MMP-9 immunohistochemistry in tumor sections showed a similar trend with a significant difference between the shCont tumors and those with ILEI KD and crizotinib in combination (Figs. 6H, 6I). NCI-H1993 tumors also expressed lower levels of MMP-2 protein upon ILEI KD (Figs. 6N, 6O) and this result was supported at the mRNA level, though without significance (Fig. 6P). Overall, these results show that both c-MET and ILEI cooperate for efficient MMP expression during growth of tumor xenografts and support the results from the cell-based assays.
Combined ILEI KD and crizotinib treatment increased E-cadherin membrane localization
To further compare tumor invasiveness and EMT status, E-cadherin-mediated cell-cell adhesion was investigated. In both, NCI-H441 and NCI-H1993 xenografts immunohistochemistry of tumor sections showed a slight increase of E-cadherin at the membranes of tumors treated with crizotinib and those derived from ILEI KD cells, and this became significant when they were in combination (Fig. 7A,B,F,G). Similar to their in vitro behavior, none of the two xenografts showed regulation of E-cadherin expression at the mRNA level upon different conditions (Fig. 7C,H), nor E-cadherin levels of tumor protein extracts showed a uniform trend of regulation (Fig. 7D,E,I,J). Therefore, these results suggest that ILEI and c-MET mainly cooperate to reduce E-cadherin protein localization at the membrane to decrease cell-cell adhesion and increase the potential for invasion.
{kind=link}
{kind=link}
{kind=link}
{kind=link}