Cell isolation and culture
Human bone marrow (BM)-MSCs were characterized by their immunophenotype CD11b-/CD19-/CD34-/CD45-/CD73+/CD90+/CD105+ and trilineage differentiation potential as previously described and used before passage [18]. They were maintained in proliferative medium consisting in α-MEM (Lonza; Levallois-Perret, France), 1 ng/mL of basic fibroblast growth factor (bFGF; R&D Systems; Noyal Châtillon sur Seiche, France), 100 µg/mL penicillin/streptomycin (PS; Lonza), 2 mM glutamine (Lonza) and supplemented with 10% fetal calf serum (FCS). Cartilage slices (<1 mm3) were recovered from the knee joints of osteoarthritic (OA) patients and incubated in 2.5 mg/mL pronase (Sigma-Aldrich, Saint-Quentin-Fallavier, France) at 37°C for 1 h followed by an incubation with 2 mg/mL type II collagenase (Sigma) at 37°C overnight. Digested cartilage tissues were filtrated through a 70 μm cell strainer and chondrocytes were seeded at 25,000 cells/cm2 in DMEM high glucose (Lonza) supplemented with 2 mmol/mL glutamine, 100 U/mL PS, 1.25 µg/mL Amphotericin B, 5 µg/mL insulin, 5 ng/mL bFGF and 10% FCS until the end of passage 0.
For proliferation assays, BM-MSCs were plated at 10,000 cells/well in 6-well plates in proliferative medium. After 5 days of culture, viable cells were counted after Trypan blue exclusion using a Malassez hemocytometer.
Differentiation assays
Chondrogenic differentiation of BM-MSCs was induced by culture in micropellets for 21 days. Briefly, 250,000 cells were pelleted by centrifugation in 15 mL conical tubes (6-8 tubes) and cultured in 3D conditions consisting in DMEM supplemented with 100 μg/mL PS, 0.35 mM proline, 0.1 μM dexamethasone, 0.17 mM ascorbic acid-2-phosphate, 1 mM pyruvate sodium, 1% insulin-transferrin-selenic acid (Lonza). In chondrogenic conditions, TGFβ3 (10 ng/mL; R&D Systems) was added at each medium change. For RNA extraction, micropellets were lyzed in RLT buffer according to the recommendations of the supplier (Qiagen; Les Ulis, France) at different time points (day 0.5, 1, 2, 3, 7, 14 and 21) and stored at -80°C.
For osteoblastogenesis, BM-MSCs were plated at 3,000 cells/cm² and cultured in osteogenic medium (DMEM containing 10% FCS, 10 mM β-glycerophosphate, 0.1 μM dexamethasone, 70 μM ascorbic acid-2-phosphate and 100 μg/mL PS). Cells were either lyzed in RLT buffer (Qiagen) at different time points (day 0.5, 1, 2, 3, 7, 14 and 21) and stored at -80°C until RNA extraction or fixed with 95% ethanol for Alizarin Red S staining at day 21.
For adipogenic differentiation, BM-MSCs were plated at 9,000 cells/cm² and cultured in proliferative medium for 5 days. The differentiation medium (DMEM-F12 containing 5% FCS, 100 μg/mL PS, 16 μM biotin, 18 μM panthotenic acid, 100 μM ascorbic acid, 60 μM indomethacin, 450 μM isobutylmethylxanthine, 1 μM dexamethasone, 1 μM rosiglitazone) was then added and changed every 3 days until day 21. Cells were either lyzed in RLT buffer at different time points (day 0.5, 1, 2, 3, 7, 14 and 21) and stored at -80°C until RNA extraction or fixed with 2.5% glutaraldehyde for Oil Red O staining at day 21.
RNA isolation and microarray hybridation
Total RNA was extracted using the RNeasy mini kit (Qiagen) following the supplier’s recommendations. RNA concentration was measured using a Nanodrop (ThermoFisher Scientific; Illkirch, France) and their integrity by using the Agilent 2100 Bioanalyzer (Agilent Technologies). Total RNA (150-200 ng) was reverse-transcribed after ribosomal fraction depletion from 2 µg total RNA, then hybridized onto GeneChip Human Exon 1.0 ST Affymetrix microarray. Ambion WT Expression Kits were used for amplification and Affymetrix WT Terminal Labeling Kits were used for labeling.
Data processing and gene expression profile analysis
After image processing with the Affymetrix GeneChip® Command Console® Software, the CEL files were analyzed using the Affymetrix Expression Console™ software v1.3.1 to obtain an intensity value signal for each probe set. The probesets were annotated using the Affymetrix annotation file from Netaffx (http://www.netaffx.com). Affymetrix array core probeset data was normalized with the Robust Multiarray Average (RMA) method. Transcripts with significant differential expression profiles were identified using the Significance Analysis of Microarray (SAM) algorithm (http://www-stat.stanford.edu/~tibs/SAM/) with the Wilcoxon test and sample label permutations (n=300) was used to identify genes of which expression varied significantly between one selected time point (day 0.5, 1, 2 or 3) and day 0. Only transcripts with a fold change (FC) ≥2 and significant false discovery rate (FDR) <5% were retained. Hierarchical clustering was produced using the Cluster and Treeview softwares (PMID: 9843981). The gene ontology (GO) enrichment analysis, the biological processes and networks of differentially expressed genes were analyzed through the use of Ingenuity Pathway Analysis (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis) (PMID: 24336805). Analysis of the expression of stemness and proliferation-related genes are based on a previously published data set from pluripotent stem cells in which a consensus stemness gene list (n=1076 genes) was defined (PMID: 17204602) and from proliferating samples including rapidly dividing CD105+ endothelial cells, CD71+ early erythroid progenitors and cell lines originating from tumors in which a proliferation gene list was defined (PMID: 19128516). Venn diagrams were obtained using Venny tools and represent the number of genes in each comparison and the overlaps between the four comparison groups.
Microarray data are in accordance to Minimum Information About a Microarray Experiment (MIAME) guidelines.
RT-qPCR analysis
RNA was reverse transcribed using the Moloney Murine Leukaemia Virus Reverse Transcriptase (Invitrogen, ThermoFisher Scientific) in a non-gradient thermocycler. qPCR was then performed on 10 ng of cDNA using SYBR™ Green I PCR Master Mix (Roche Diagnostics, Meylan, France) or TaqMan™ Universal Master Mix II, with UNG and specific primers (Tables 1-2). PCR reaction was performed as follow: 95°C for 5 min; 40 cycles at 95°C for 15 s; 64°C for 10 s and 72°C for 20 s with SYBR Green or 95°C for 5 min; 40 cycles at 95°C for 15 s and 64°C for 10 s with TaqMan, in a ViiA 7 Real-Time PCR System (Applied Biosystems, ThermoFisher Scientific) and analyzed with the dedicated software. All values were normalized to the housekeeping gene RPS9 and expressed as relative expression using the formulae 2−∆Ct or as fold change using the formulae 2−∆∆Ct.
Cell transfection
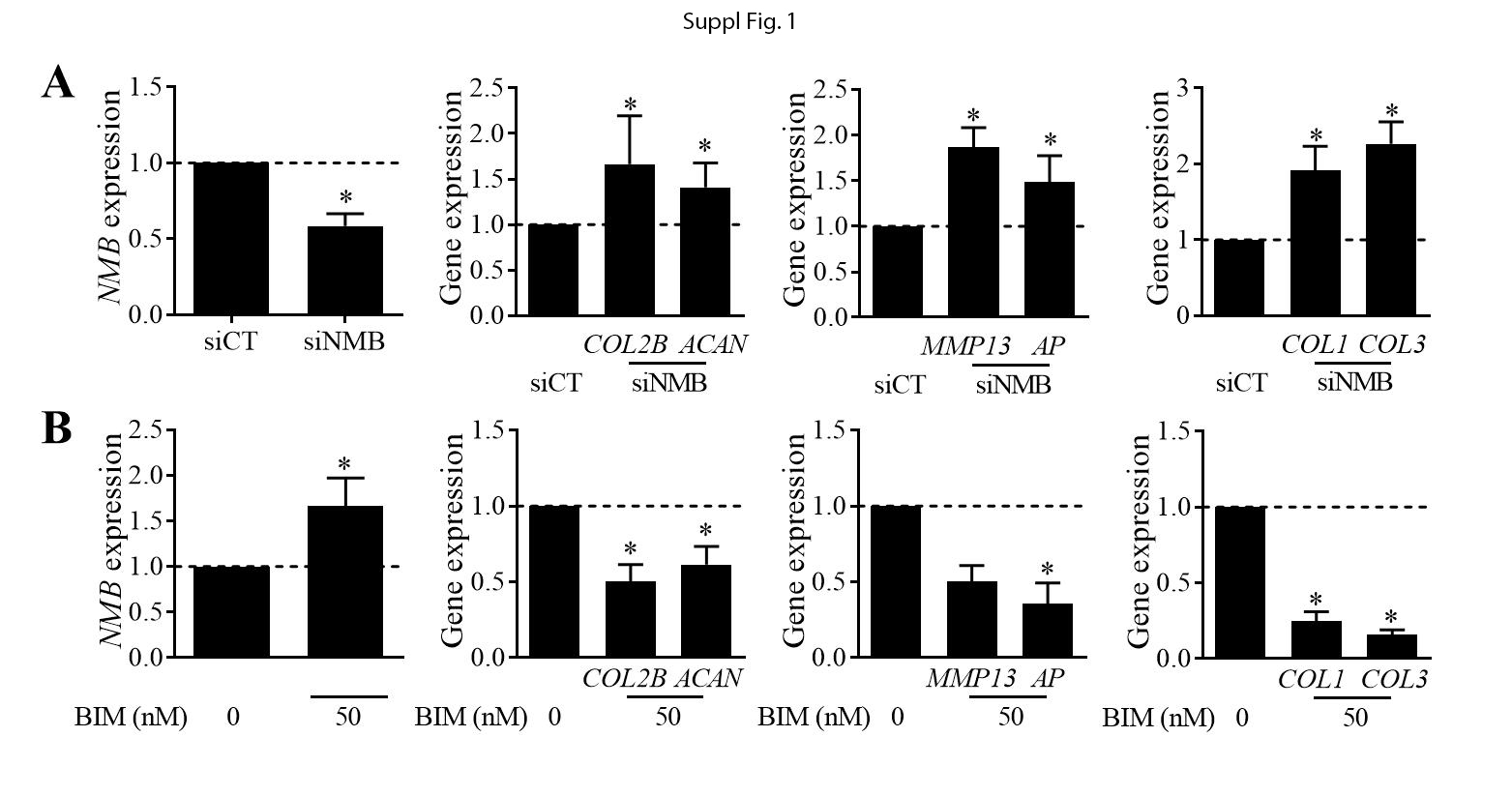
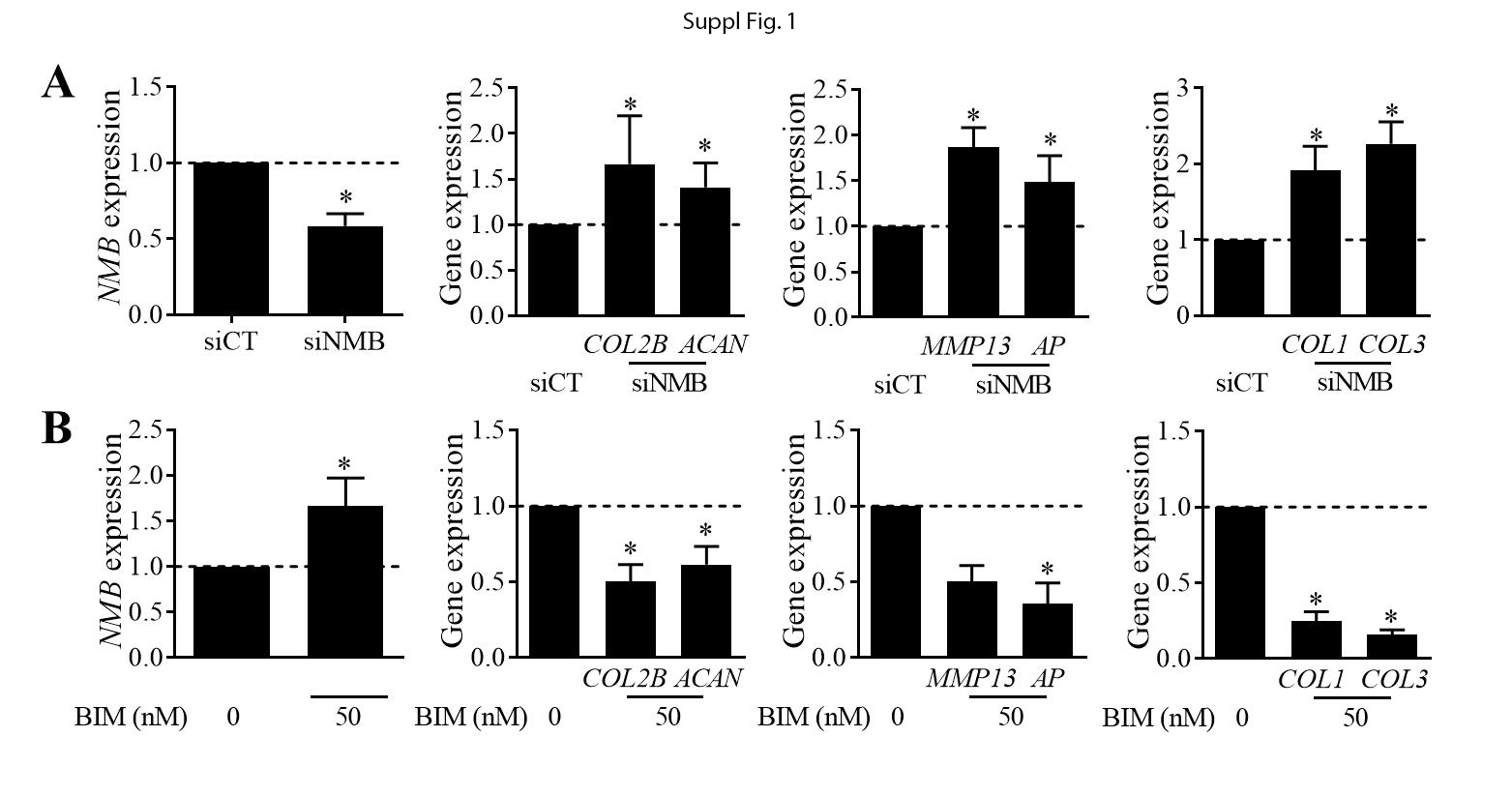
BM-MSCs were transfected twice: 4 days (80% of confluence) and one day before inducing the chondrogenic differentiation. Transfection was done using 50 nM of predesigned siNMB (TaqMan, siRNA ID: s9582; Applied BIosystems, Illkirch, France) or siRNA control (UAAGGCUAUGAAGAGAUACTT) using Oligofectamine™ in Opti-MEM™ (Life Technologies SAS, Villebon-sur-Yvette, France) during 6 hours.
Cell treatment
BM-MSCs were treated with recombinant NMB (rNMB; peptide sequence: Gly-Asn-Leu-Trp-Ala-Thr-Gly-His-Phe-Mel-NH2; Sigma-Aldrich) for the 21 days of chondrogenesis. Two concentrations were evaluated: 100 or 1000 nM and the medium was changed every 3-4 days. Passage 1 chondrocytes were plated at 60,000 cells/well of 6-well plates and treated with the NMBR antagonist BIM23042 (BIM; peptide sequence: D-Nal-Cys-Tyr-D-Trp-Lys-Val-Cys-Nal-NH2; Tocris, Noyal Châtillon sur Seiche) at 50 nM during 3 days.
Western blot
Micropellets were first washed with PBS, then stored in RIPA lysis buffer (Sigma Aldrich) containing a protease inhibitor cocktail (Sigma, P-8340) for 1h at -80°C. Micropellets were then grinded and centrifuged at 12,000 rcf, at 4°C for 15 min. Total proteins (50 µg) diluted in Bolt LDS sample buffer and Bolt Sample Reducing Agent were heated at 70°C for 10 min and loaded on Bolt 4-12% Bis-Tris Plus precast gels (Life Technologies). Samples were allowed to migrate for 30 min at 200 mV using the MOPS SDS running buffer and transferred on nitrocellulose membranes using the iBlot 2® Gel Transfer Device (Life Technologies). Membranes were then blocked with 5% milk and probed with the anti-human NMB rabbit polyclonal primary antibodies (1:500; Cohesion Biosciences, Nanterre) at 4°C, overnight. Anti-human Actin mouse polyclonal primary antibodies (1:5000) for 2h at room temperature. After several rinses with tris phosphate buffered saline (TBS) containing 0.05% tween-20 (Sigma-Aldrich), membranes were incubated with a horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:50000) for 1h at room temperature. Blots were revealed using a WesternBright™ Sirius Chemiluminescent detection kit (Advansta, Blagnac) and scanned (ChemiDoc XRS System, Biorad).
Analysis of spontaneous calcium transients and calcineurin cellular activity
Spontaneous calcium transients were monitored on micropellets at day 3 of differentiation. Micropellets were rinsed with buffer containing 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 10 mM glucose (pH 7.4) and incubated with Fluo 4-AM (500 µg/mL; Invitrogen) and Pluronic F-127 (0.1%; Invitrogen) at 37°C for 45 min. Calcium imaging was performed for 30 min using a Multi-photon Zeiss LSM 7MP OPO microscope (Zeiss, Marly le Roi, France) and analyzed with the ZEN software (Zeiss).
Calcineurin activity was measured using the calcineurin cellular activity assay kit (Enzo, Villeurbane, France). Day 3-micropellets were rinsed in Tris Buffered Saline (TBS), lysed in the lysis buffer containing the protease inhibitor cocktail and grinded using an Ultra-turrax dispenser. Samples were then stored at -80°C before quantification of calcineurin cellular activity by measuring the Ca2+/calmodulin-dependent Ser/Thr protein phosphatase 2B (PP2B) activity according to manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 software (San Diego, USA). Data were represented as mean ± standard error of the mean (sem) of separate experiments. Data did not assume a Gaussian distribution and were analyzed using appropriate nonparametric statistical tests. The comparison between 2 unpaired groups was performed using a Mann-Whitney test. The comparison between one group compared to the control group normalized to 1 or 100% was performed using a Wilcoxon signed rank test. The comparison between different groups was analyzed by a Kruskall-Wallis test followed by a Dunn’s multiple comparisons test. Correlations were analyzed with a nonparametric Spearman test. Statistical differences were indicated as *: p < 0.05, **: p < 0.01, ***: p < 0.001.
{kind=link}
{kind=link}