2.1. Materials: Scutellaria barbata was purchased from Hebei Jinyezi Pharmaceutical Co., Ltd. (Hebei, China); the finely sieved powder was kept in a constant weight dryer until it was dry. The fine powder was crushed through a 40 mesh sieve and stored in a dryer until use. DEAE-52 cellulose was purchased from Phygene Biotechnology Co., Ltd. (Fuzhou, China), and Sephadex G-100 was purchased from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). Monosaccharide standards were provided by Shenzhen Bo Rui Sugar Biology Co., Ltd. (Shenzhen, China). All other reagents used in the tests were analytical grade and obtained from local chemical suppliers in China. The human hepatoma HepG2 cell line was purchased from the Chinese Academy of Sciences (Shanghai, China). MEM dry powder, penicillin and streptomycin, trypsin, non-essential amino acids (NEAA) and sodium pyruvate in 100 µg/ml penicillin and 100 µg/ml streptomycin were purchased from Gibco Inc. (Gibco, USA). Australian fetal bovine serum (FBS) was purchased from Wuxi Xinrun Biotechnology Co., Ltd. (Wuxi, Jiangsu). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation and cytotoxicity test kits and DNA content test kits were purchased from Solebao (Beijing, China). A Hoechst 33258 apoptosis staining kit was purchased from Beyotime Co., Ltd. (Nanjing, China).
2.2. Optimization of water extraction and alcohol precipitation with response surface methodology
2.2.1 Single factor experimental design
Chunlin Ye optimized the extraction parameters for Scutellaria barbata with response surface methodology, and the polysaccharide yield was only 2.43 ± 0.11%(26). To further improve the polysaccharide yield based on the process for extraction of crude Scutellaria barbata polysaccharide, we selected three factors: liquid-material ratio, extraction temperature and extraction time. Five levels were investigated for each factor(27, 28), including: liquid-material ratios; 1:10, 1:15, 1:20, 1:25 and 1:30; extraction temperatures: 60 ℃, 70 ℃, 80 ℃, 90 ℃ and 100 ℃; and extraction times: 1 h, 2 h, 3 h, 4 h and 5 h. The content of SBP was determined.
2.2.2 Response surface methodology
According to the results of the single-factor experiments, three factors and five levels of box Behnken experimental design were carried out to optimize the extraction conditions, and the related experiments were generated by Design Expert 11 (State-Ease Inc., Minneapolis, MN, USA) RSM software and repeated three times for each group. RSM was used for regression analyses of the experimental data, and a nonlinear quadratic model was fitted according to the formula(29, 30).
2.3. Extraction of Scutellaria barbata crude polysaccharide (SBP) by water extraction and alcohol precipitation
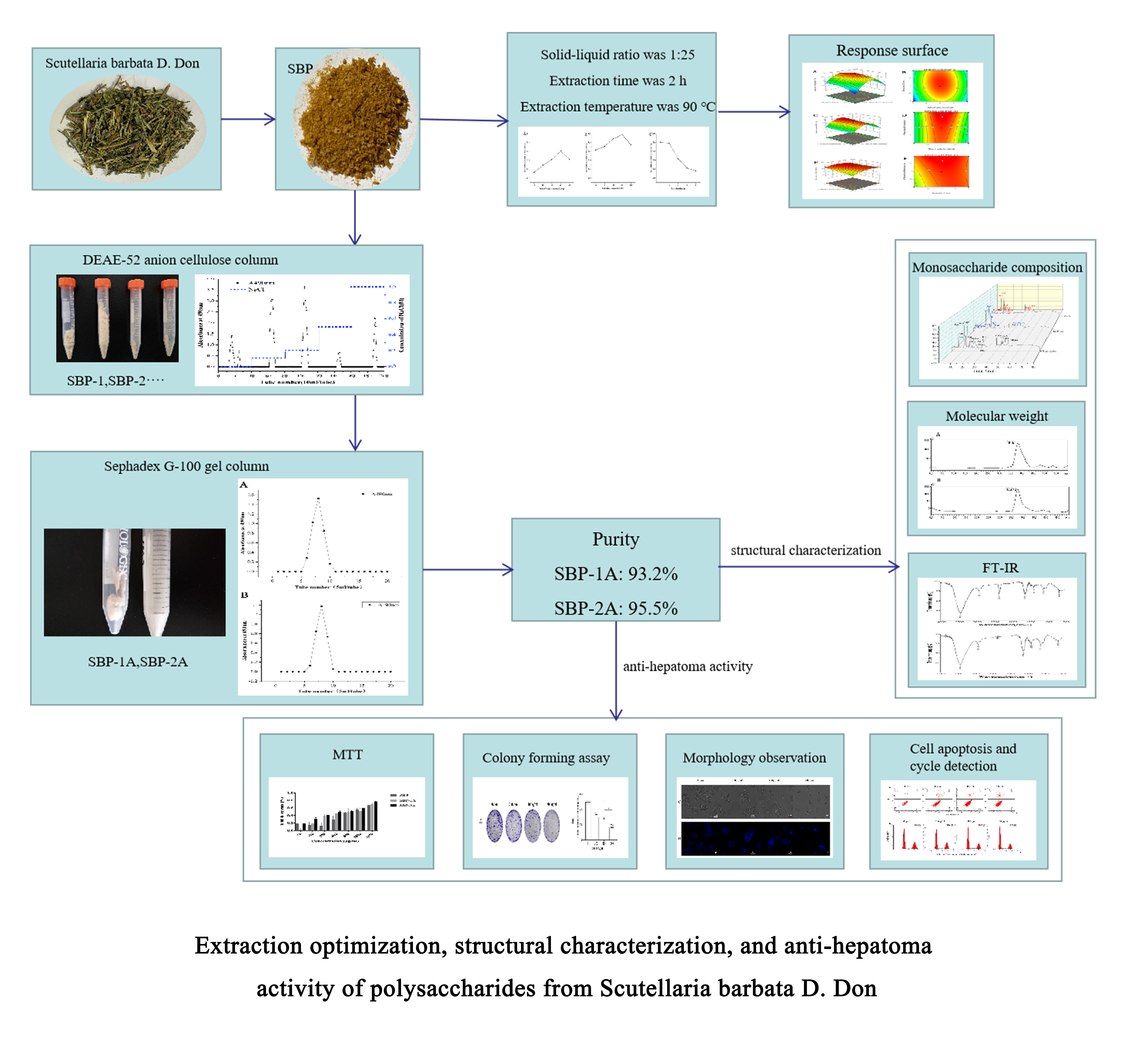
Scutellaria barbata powder was soaked in 95% ethanol solution several times to decolorize the powder and remove alcohol-soluble components. The filter residue was dried for 24 h to prepare the Scutellaria barbata sample. With a ratio for material to liquid of 1:25 (g/ml), 90 ℃ hot water extraction was used for two extractions lasting 2 h each time, the mixture was centrifuged for 15 min at 3500 rpm/min, the supernatant was combined, rotated and evaporated at 55 °C and concentrated to 1/4 of the original volume; 4 volumes of absolute ethanol were added slowly to the concentrated solution with stirring, and the solution was cooled to 4 ℃. The precipitate was collected, diluted with water and deproteinized (volume ratio 1:5) with Savage reagent (the chloroform:n-butanol volume ratio was 4:1)(31, 32). The solution was decolorized with macroporous resin D101 and filtered, and the extract was separated and concentrated to 1/4 of the original volume under reduced pressure at 55 °C. SBP was prepared by vacuum drying at 50 °C and stored in a dryer until use.
2.4. SBP purification
DEAE-52 cellulose was loaded onto an ion chromatography column (2.5 cm × 100 cm) for purification of polysaccharides with an ultrapure water balance. Polysaccharide (200 mg) was dissolved in 10 ml of distilled water, injected into the balanced chromatographic column, eluted successively with distilled water, 0.1 M, 0.2 M, 0.3 M, 0.5 M and 1 M NaCl solution gradient. The flow rate was adjusted to 1 ml/min, 10 ml/tube of eluent was collected by the automatic collector, and the absorbance at 490 nm was measured with the phenol sulfuric acid method(33). The elution solutions were combined, rotated and concentrated to 1/8 of the original volume at 55 ℃; a dialysis bag with molecular weight of 3500 Da was used for 24 h to collect the dialysate, which was prefrozen at -40 ℃ for 24 h, and then freeze-dried into powder(34). Samples were placed on a SephadexG-100 dextran gel column and washed with ultrapure water at 5 ml/tube. The polysaccharide content of each tube was measured by the phenol sulfuric acid method, and elution curves were drawn. If the component showed a single symmetrical elution peak after the SephadexG-100 gel column, it contained a single polysaccharide component. The main peak components were concentrated, frozen and dried to obtain white flocculated polysaccharides.
2.5. Polysaccharide and protein content
A glucose standard solution, 0.1 mg/ml, was prepared and 0, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, and 1.8 ml of glucose standard solution was added into stoppered test tubes. Distilled water was added to establish a volume of 2 ml, 1 ml of 5% phenol was added and mixed well, and 5 ml of concentrated sulfuric acid was added quickly. The mixture was warmed in a 40 ℃ water bath for 30 min and allowed to stand for 20 min, after cooling to room temperature, the absorbance value at 490 nm was measured with an ultraviolet spectrophotometer; the glucose standard concentration (μg/ml) was the abscissa, and the absorbance value A was the ordinate used to draw the glucose standard curve. One milligram of SBP was weighed and fully dissolved in distilled water. The absorbance value was determined at 490 nm according to the phenol sulfuric acid method(33), and the polysaccharide concentration in the sample was calculated according to the standard curve. Then, according to the number of standards and samples, a BCA working solution was prepared to give a 50:1 ratio of BCA reagent and Cu reagent. A 10 μl sample of BSA standard was diluted to 100 µl (0.5 mg/ml) with PBS. The standard (0, 2, 4, 6, 8, 12, 16, 20 μl) was added to 96 well plates, and PBS was added to increase the volumes to 20 μl. The sample was weighed and diluted with PBS, and 20 µl was added to a 96 well plate with 200 µl/well BCA working solution; the plate was warmed at 37 ℃ for 15-30 min. The absorbance value at 562 nm was measured by enzyme labeling instrument (Multiskan MK3, ThermoFisher, USA), and the protein concentration was calculated according to the standard curve.
2.6. Monosaccharide composition
Sixteen monosaccharide standards were prepared as 10 mg/ml standard solutions. The 0.01, 0.1, 0.5, 1, 5, 10 and 20 mg/L gradient concentration standards of monosaccharide standard solutions were labelled as standards 1-7. Ten milligrams of sample was accurately weighed and placed into an ampoule bottle, 10 ml of 3 M TFA was added and the substrate was hydrolyzed at 120 ℃ for 3 h. The acid hydrolysis solution was absorbed, transferred into the tube, blown dry with nitrogen, added to 5 ml of water, vortexed and mixed evenly, diluted to 100 µl, added to 900 µl of deionized water, and centrifuged at 12,000 rpm for 5 min. The supernatant was removed for ion chromatography (IC) analysis(35) with an ICS5000 system (Thermo Fisher, USA).
2.7. Molecular weight determination
High-performance liquid chromatography (HPLC) is widely used in determinations of molecular weight because of its high accuracy and efficiency(36). Samples and standards were weighed accurately (molecular weights were 5000, 11600, 2800, 48600, 80900, 148000, 273000, 409800, 667800 and 3693000 Da, respectively), 5 mg/ml solutions were prepared, the solutions were centrifuged at 12000 rpm for 10 min and the supernatant was filtered through 0.22 μm microporous filters, and the sample was transferred into a 1.8 ml injection vial. The chromatographic conditions for HPLC (LC-10A, Shimadzu, Japan) were as follows: chromatographic column: BRT105-104-102 tandem gel column (8×300 mm); mobile phase: 0.05 NaCl solution; flow rate: 0.6 ml/min, column temperature: 40 ℃; injection volume: 20 μl; detector: differential refractive index detector RID-1OA. By taking the logarithm of the molecular weight of the standard (log(Mw)) as the ordinate and the peak time (min) as the abscissa, regression fitting of the curve was performed with software to obtain the standard curve for molecular weight distribution. The chromatogram of the sample was obtained with the chromatographic separation conditions described above, the retention time of a single symmetrical peak was recorded, and the molecular weight was calculated.
2.8. FT-IR analysis
Samples were analyzed by Fourier transform infrared (FT-IR) spectroscopy(37) in a pressed KBr pellet. Polysaccharide (5 mg) was weighed, put into a mortar with dried KBr, mixed and ground fully, pressed into a thin sheet with a pellet press, and analyzed with a Thermo Scientific Nicolet iS5 FT-IR spectrophotometer (Thermo Nicolet Co., Madison, WI, USA) with a scan range of 500-4000 cm-1.
2.9. Anti-hepatoma activity
2.9.1 MTT assay
The MTT assay was used to screen the SBP component with the highest antitumor activity. HepG2 cells were inoculated in a 96 well microplate, warmed in an incubator to 37 ℃ under 5% CO2 to adhere to the wall, and cultured with different concentrations (50-3200 μg/ml) of SBP, SBP-1A and SBP-2A for 48 h. Then 20 µl/well of MTT solution (5 mg/ml) was added, the mixture was cultured for 4 h, the liquid was discarded and 150 µl/well of DMSO was added. The system was oscillated at low speed for 10 min, and the absorbance (OD) value at 490 nm was measured with a SpectraMax M3 microplate reader (Molecular Devices, USA). A1 represents the OD value of the experimental group, and A2 represents the OD value of the negative control group.
Inhibition rate (%) = (1-A1/A2) × 100%
The IC50 values of three polysaccharides with different concentrations were calculated by GraphPad Prism software. According to the IC50 value, low, medium and high doses of SBP-2A were determined for studying the anti-hepatoma effect.
2.9.2 Colony forming assay
HepG2 cells were added at 4000 cells/well and inoculated in 35 mm dishes, adherent HepG2 cells were treated with SBP-2A for 48 h. The same amount of MEM complete medium was added to each dish for routine culturing for 10 days. The culture medium was discarded and the cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min. The paraformaldehyde was discarded and the cells were washed twice with PBS, dyed with 0.1% crystal violet for 20 min, cleaned with PBS buffer and photographed, and the number of colonies with more than 50 cells were counted. The colony forming ability of HepG2 cells treated with different concentrations of SBP-2A was detected by a cell colony forming assay.
Clone formation rate (%) = number of cell clones/number of inoculated cells.
2.9.3 Cell morphology observation
HepG2 cells in the logarithmic growth stage were cocultured with SBP-2A for 48 h. The morphological changes in HepG2 cells were directly observed with a DMI 6000B Leica microsystem (Wetzlar, Germany). Then, the culture medium was absorbed, 0.5 ml of fixing solution was added, and the cells were fixed for 20 min; the cells were washed with PBS, cultured with Hoechst 33258 staining solution for 10 min, and rinsed with PBS. After blocking with an anti-fluorescence quencher, morphological changes due to apoptosis of HepG2 cells induced by SBP-2A were observed under a DMI 6000B Leica microsystem.
2.9.4 Annexin V-FITC/PI staining assay and cell cycle detection
HepG2 cells were added at 5 × 105 cells/well and inoculated in 35 mm dishes, and the cells were treated with SBP-2A for 48 h. The cells were digested, centrifuged (700 rpm, 4 min), collected and washed twice with precooled PBS to obtain the cell precipitate, which was resuspended in 500 μl of binding buffer. Then, 5 μl Annexin V-FITC dye solution and 10 µl PI were added with mixing, and the reaction was kept in the dark at room temperature for 15 min. Finally, apoptosis was evaluated by a Cytomics FC500 Flow Cytometry CXP system (Beckman, USA), and the percentage of apoptotic cells was analyzed by FlowJo v10.6.2. software.
The cell cycle distribution of the SBP-2A-treated HepG2 cell line was evaluated by flow cytometry. The cells were seeded at a density of 5×105 cells/well and inoculated in 35 mm cell culture dishes. After adding adherent, the cells were starved with MEM culture medium containing 1% FBS for 12 h. The cells were treated with SBP-2A solutions for 48 h. With a sample collection rate of 1×106 cells/piece, cell precipitates were obtained by centrifugation, 300 µl of precooled PBS was added to resuspend them, 700 µl of precooled absolute ethanol was slowly added drop by drop with mixing, and then the cells were fixed overnight at -4 ℃. The fixing solution was washed away with PBS, and 100 µl of RNase A solution was added to the cell precipitate to resuspend it. The mixture was warmed in a water bath at 37 ℃ for 30 min, 400 µl of PI staining solution was added and mixed, and the cells were incubated in the dark at 4 ℃ for 30 min. Finally, the cell cycle of the sample was evaluated by Cytomics FC500 Flow Cytometry CXP (Beckman, USA) and the cycle results were analyzed by ModFit LT 5.0 software.
2.10 Statistical analyses
All data were expressed as the mean ± standard deviation (SD). All experiments were repeated three times. One-way ANOVA and multiple comparisons were used, and all statistical analyses were performed using GraphPad Prism 7.0 software and plotted with Origin2021. P < 0.05 was taken to indicate a statistically significant difference.
{kind=link}