Absence of miRNAs in T. vaginalis
Three T. vaginalis strains were employed for deeply sequencing the 18-40 nt small RNAs. As showed in Table 1, the three libraries yielded 6 303 530, 7 071 840, and 5 809 794 reads that perfectly mapped to the T. vaginalis genome, respectively, accounting for an average of 91.76% of the clean data. The distribution of mapped reads from these three isolates was relatively consistent, all displaying three main peaks at the size 29, 32 and 36 nt (Fig. 1a). Therefore, these data were pooled together for further analysis to enhance the identification of novel type of small RNAs. Apart from the three key peaks as expected in total reads, a single peak at 34 nt showed in unique reads (Fig. 1b). Such feature was incredibly different from those of model organisms, which generally demonstrated a peak at ~22 nt dominated by miRNAs in both total and unique read aspects [29, 30, 31]. This suggested that the composition of small RNAs in T. vaginalis might be divergent.
The data were then annotated and the abundance of each type of small RNAs was assessed for its expression level. As illustrated in Fig. 1c, these small RNAs were originated from ribosomal RNAs (rRNAs), tRNAs, messenger RNAs (mRNAs), small nuclear RNAs (snRNAs) / small nucleolar RNAs (snoRNAs), as well as unannotated small RNAs. Unexpectedly, miRNAs in our pool showed at extremely low abundance (0.0046%). Among the 27 miRNAs identified previously [23, 24, 25], only nine were detected in our deep sequencing data with particular low counts, except for one namely ‘tvm-005’ derived from tRNA (Additional file 1: Table S3). In contrast, tsRNAs predominated after rRNA-derived reads and accounted for 12.79% of clean reads. These molecules prevailed in T. vaginalis genomic contig DS113177 to DS127907, from where tRNA genes were mainly coded (Fig. 1d).
Profiles of tsRNAs fromT. vaginalis
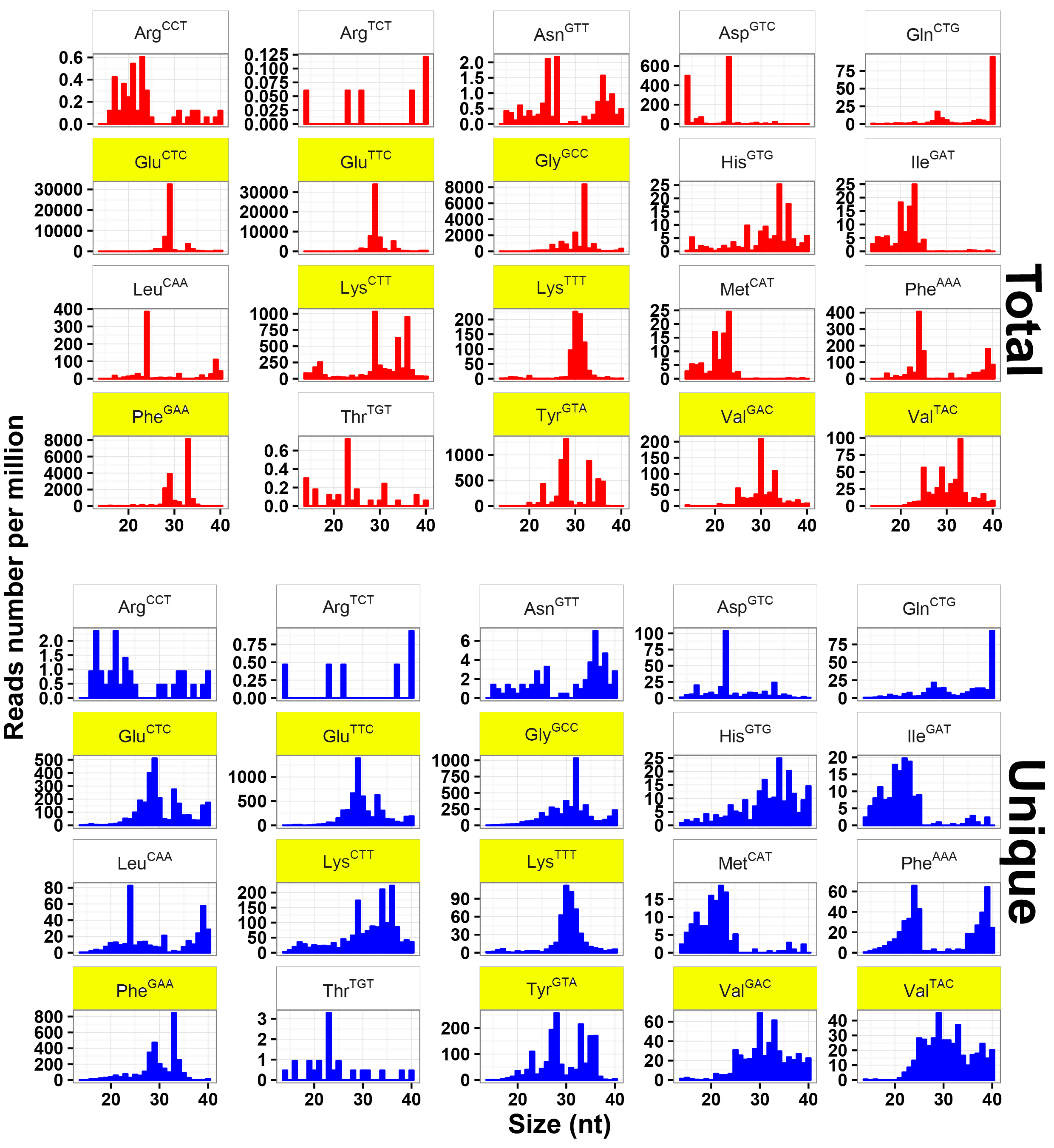
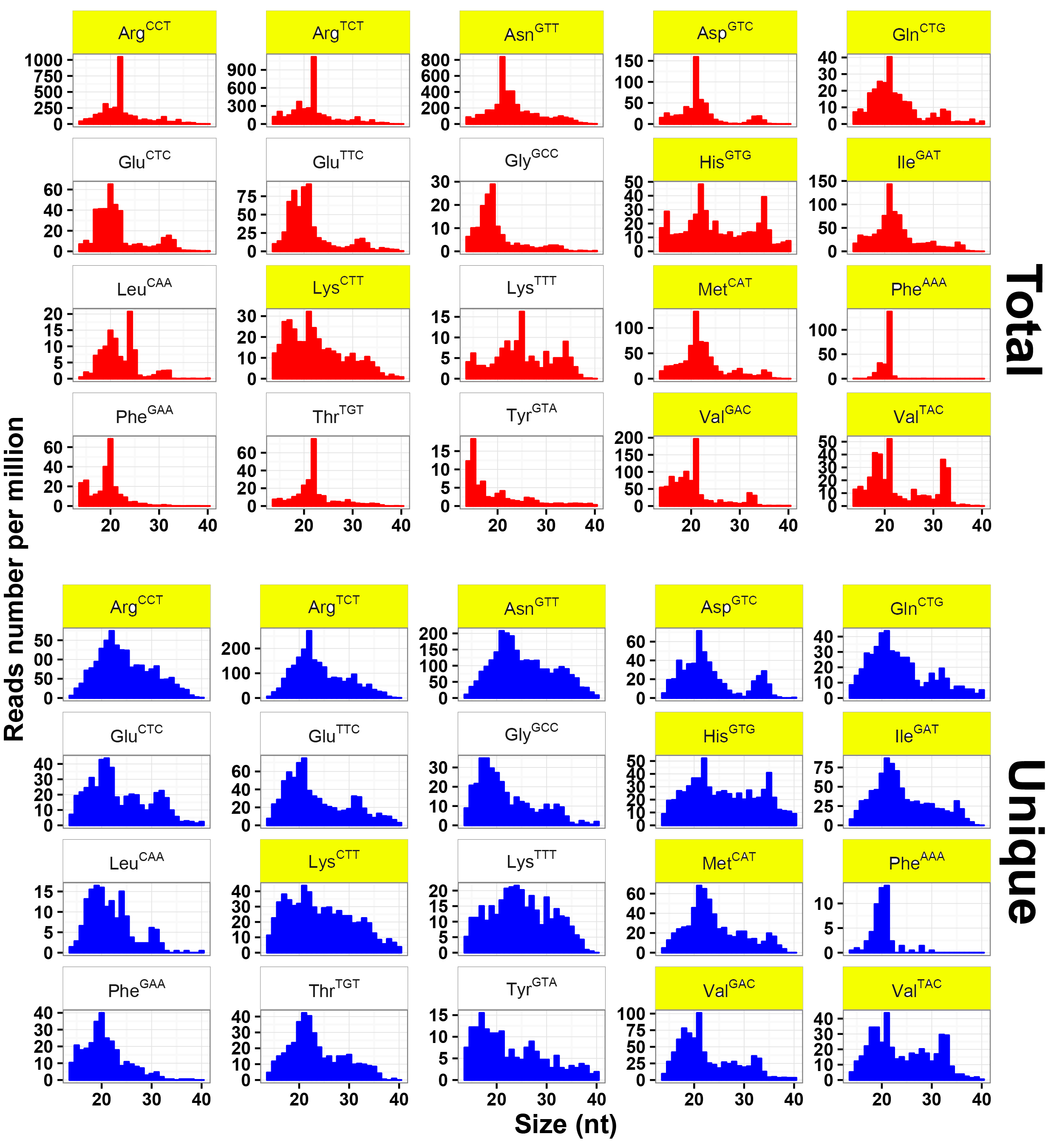
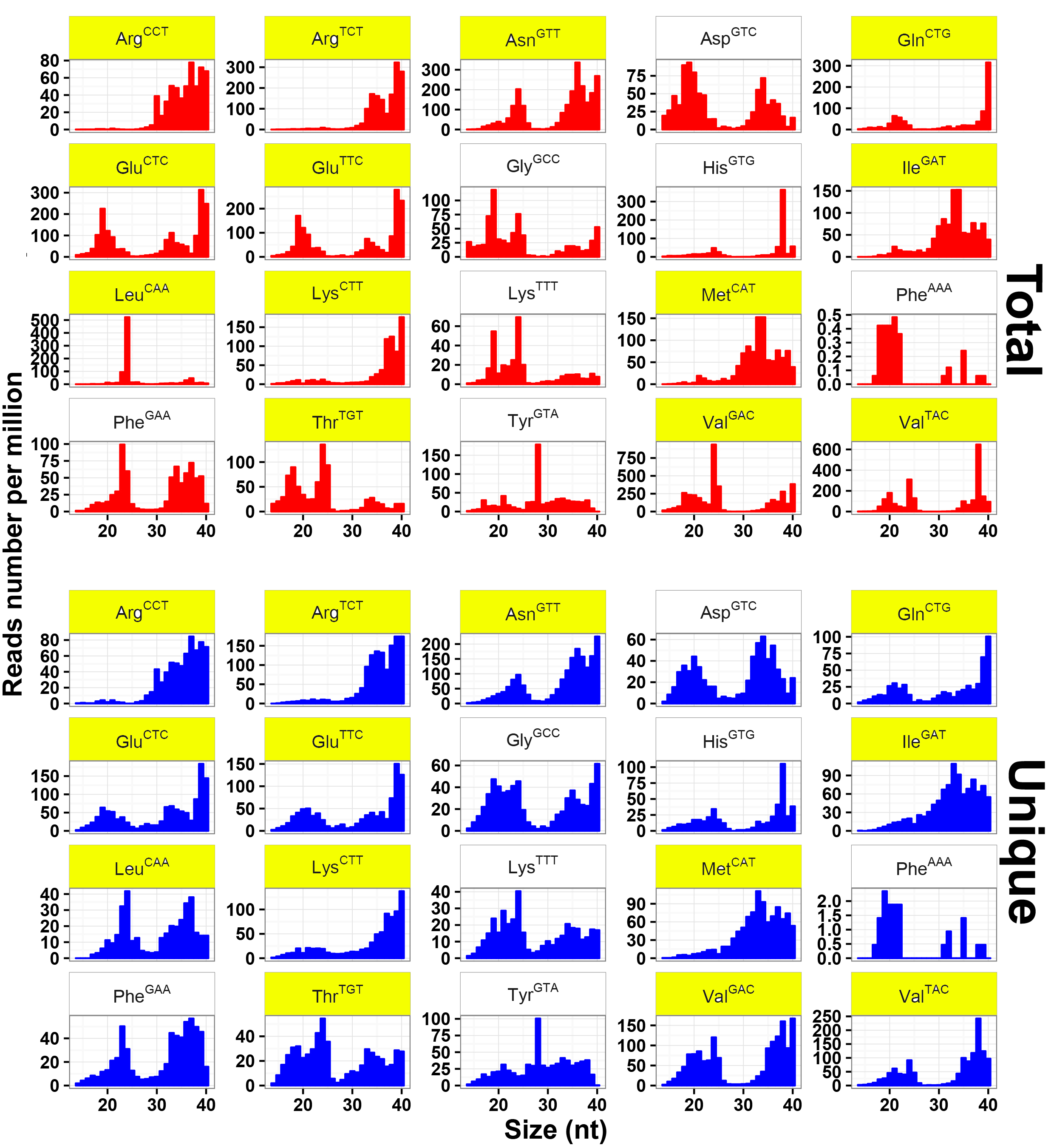
To explore the expression profiles of tsRNAs, their parental tRNAs consisting of 20 amino acids were analyzed. Eight amino acids, including Glu, Gly, Phe, Lys, Val, Arg, Asn, and Tyr, were found to produce up to 85% of tsRNAs (Fig. 2a). Such expression bias indicated that tsRNAs were not generated by random degradation of mature tRNAs. The distribution pattern of tsRNAs was further investigated. Other than that in Fig. 1b, it showed only two conspicuous peaks dominating at ~29 and ~33 nt in total tsRNAs (Fig. 2b), implying multiple types of tsRNAs with diverse sizes in T. vaginalis. These tsRNA reads were therefore mapped to all 165 Trichomonas tRNAs. As expected, their biogenesis from the parental tRNAs were found relatively conserved as previously reported [10, 19, 20, 21], with a large amount of reads aligned to three main positions at 5' end, anticodon area and 3' end of mature tRNAs (Fig. 2c). Consequently, these three types of Trichomonas tsRNAs were named as 5'tritsRNAs, mid-tritsRNAs and 3'tritsRNAs, respectively. The homogeneity of these tritsRNAs based on their types and size distributions were investigated by plotting the RPM values against sizes in both total and unique reads. Two peaks were observed for 5'tritsRNAs at ~29 nt and ~33 nt, for mid-tritsRNAs at ~21/22 nt, and for 3'tritsRNAs at ~24 nt and ~40 nt, respectively (Fig. 2d), displaying great difference in their sizes. The double peaks occurred in the plots of 5'tritsRNAs and 3'tritsRNAs suggested that there were at least two subgroups of tritsRNAs in each.
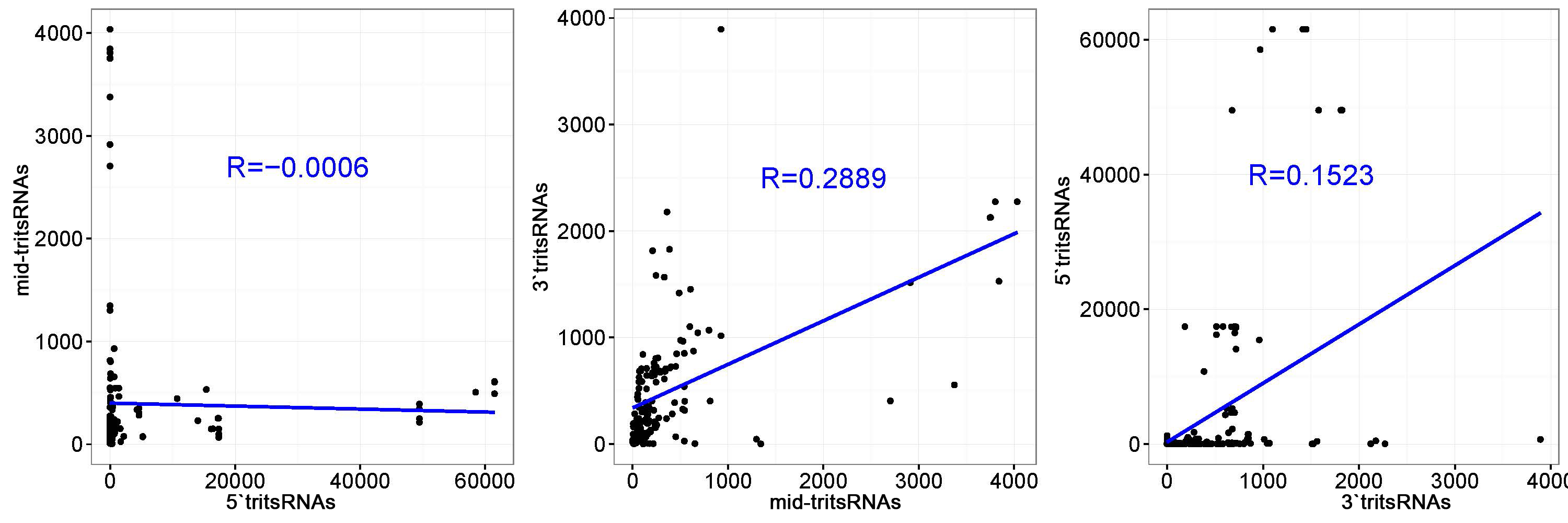
We subsequently selected the top 20 tRNAs that produced high level of tritsRNAs to further evaluate their expression divergence. Among them, seven primarily produced 5'tritsRNAs, three generated mid-tritsRNAs and five created 3'tritsRNAs, and the rest yielded tritsRNAs from multiple regions (Fig. 2c). 5'tritsRNAs prevailed (67.62%) in tritsRNAs, whereas mid-tritsRNAs and 3'tritsRNAs accounted for only 7.37% and 15.86%, respectively (Fig. 2e). No significant association between the abundance of any two types of tritsRNAs was found by Pearson correlation analysis (Additional file 2: Figure S1). The predominated tritsRNAs in each group were then examined by plotting the RPM values of all 60 tritsRNAs from these tRNAs against their sizes. A total of 32 tritsRNAs were identified to show consistent peaks as in Fig. 2d, including nine 5'tritRFs, twelve mid-tritsRNAs, and eleven 3'tritRFs, as displayed in Additional file 3, 4 and 5: Figure S2, S3, S4 and Table 2. Consequently, these 32 tritsRNAs were employed for further classification and confirmation analysis.
Identification of tritRFs and tritRNA-halves
Given that tRFs, typically shorter than 32 nt [8], are the theoretical products of tRNA-derived small RNAs under ordinary condition, our analysis identified longer products in both 3' and 5'tritsRNAs. To further classify these tritsRNAs, we investigated their cleavage sites by aligning each of those 32 tritsRNAs to its parental tRNA. As illustrated in Fig. 3, two sites were identified to produce 5'tritsRNAs, with one occurring in the anticodon loop to generate those of 33 nt, and one in the anticodon arm to yield these of 29 nt. The mid-tritsRNAs originated from two sets of combined endonucleolytic cleavages both in the arms neighboring to anticodon loop and TψC loop, however, one group covered the anticodon loop while the other involved the TψC loop. Three cleavage sites in anticodon loop, anticodon arm and TψC arm resulted in the generation of 3'tritsRNAs with sizes of 40, 33 and 24 nt, respectively. Therefore, 5'tritsRNAs of 29 nt, mid-tritsRNAs and 3'tritsRNAs of 24/33 nt were produced by combined cleavages other than in the anticodon loop and belonged to the class of tritRFs. While two types (5'tritsRNAs of 33 nt and 3'tritRNAs of 40 nt) were generated by single cleavage in the anticodon loop and thus were classified as tritRNA-halves (Table 2).
Experimental confirmation of tritsRNA candidates and motif prediction
We further experimentally evaluated the presence of tritRFs and tritRNA-halves in T. vaginalis to confirm our findings. Stem-loop RT-PCR successfully amplified 25 (nine 5'tritsRNAs, seven mid-tritsRNAs, and nine 3'tritsRNAs) of these 32 candidates, corresponding to 78.1% of tested candidates. This proved the expression of both tritRFs and tritRNA-halves in T. vaginalis transcriptome (Fig. 4a).
The motifs around the cleavage sites that might guide the generation of tritsRNAs were then explored. By employing GLAM2 algorithm, three motifs (Motif TV1-3, Fig. 4b) were successfully predicted to process tritsRNAs. Motif TV1 was located along D-stem and anticodon-arm for the 3' end cleavage of 5'tritsRNAs (29 nt). Motif TV2 and TV3 were both in TψC stem to process the 3' end cleavage of mid-tritsRNAs (22 nt) and 5' end cleavage of 3'tritsRNAs (24 nt), respectively. Albeit both motif TV2 and TV3 contained highly conservative ‘UUC’ triple-nucleotide element, they seemed to take charge of generating various types of tritsRNAs (Fig. 4b).
{kind=link}
{kind=link}
{kind=link}
{kind=link}