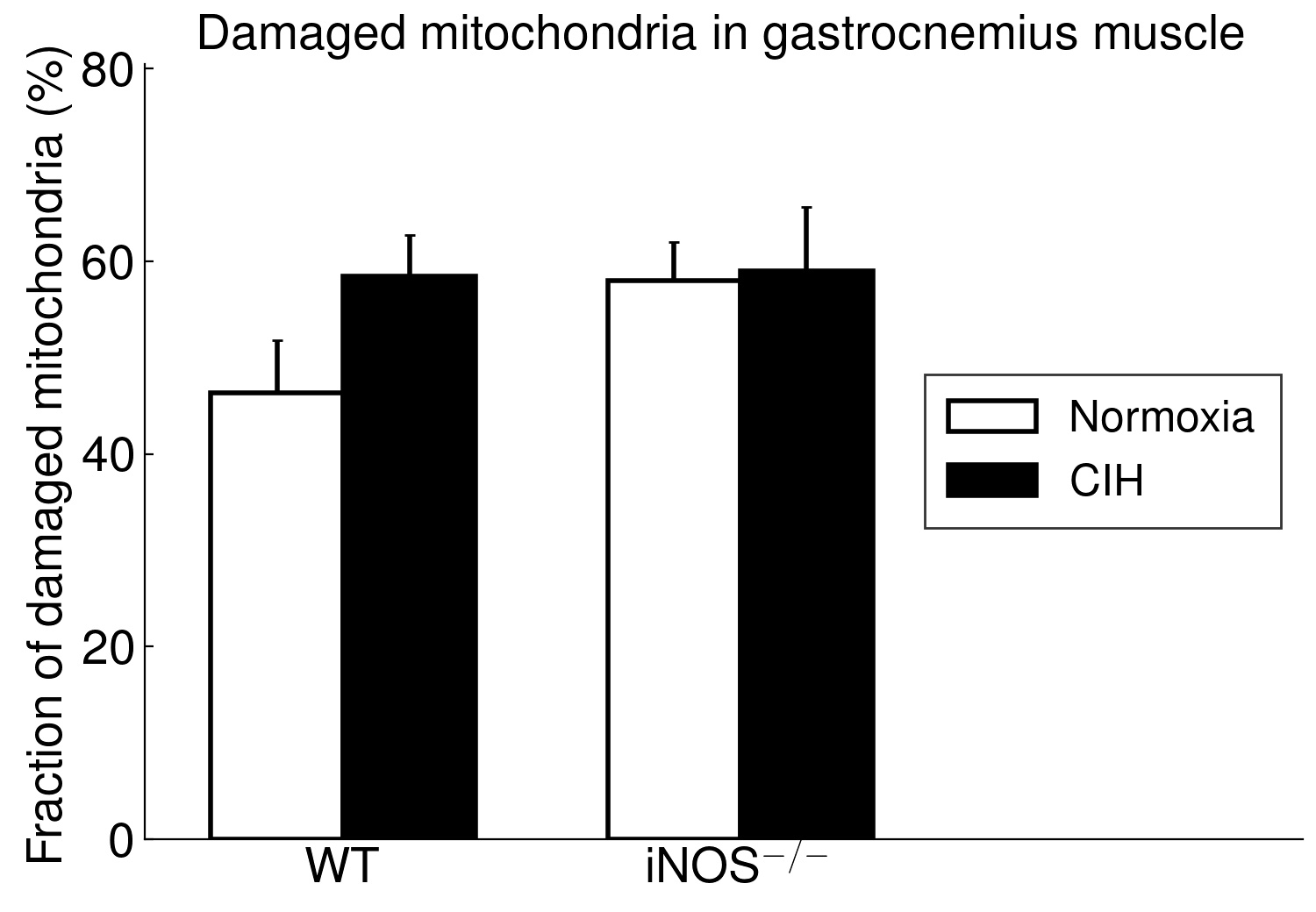
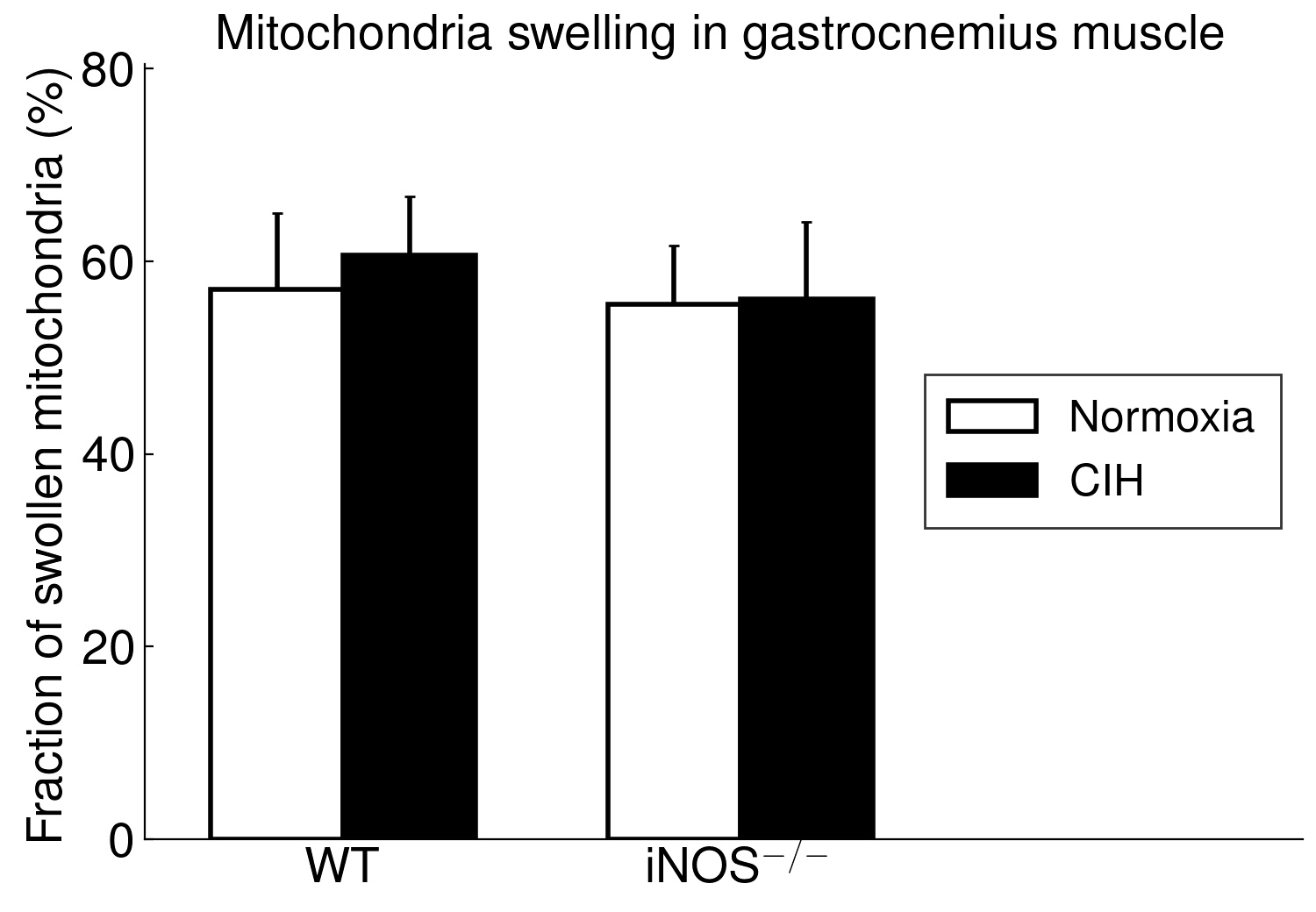
Using long-term CIH exposure in mice as a model of OSA, the present study shows for the first time that CIH compared to NOX causes damage of potential functional relevance in ‘red’ (soleus) but not in ‘white’ (gastrocnemius) muscle. This comprises a reduction in area, in length and, by trend, in integrity of postsynaptic NMJ as well as in size (CSA) and fraction of type 2a fibers (at higher type-1 fiber fraction). Moreover, these changes were associated with considerable mitochondrial damage, which showed a significant correlation to (loss in) NMJ area (r=-0.71, p < 0.001) and were, again, limited to soleus muscle, while gastrocnemius revealed no significant mitochondrial damage.
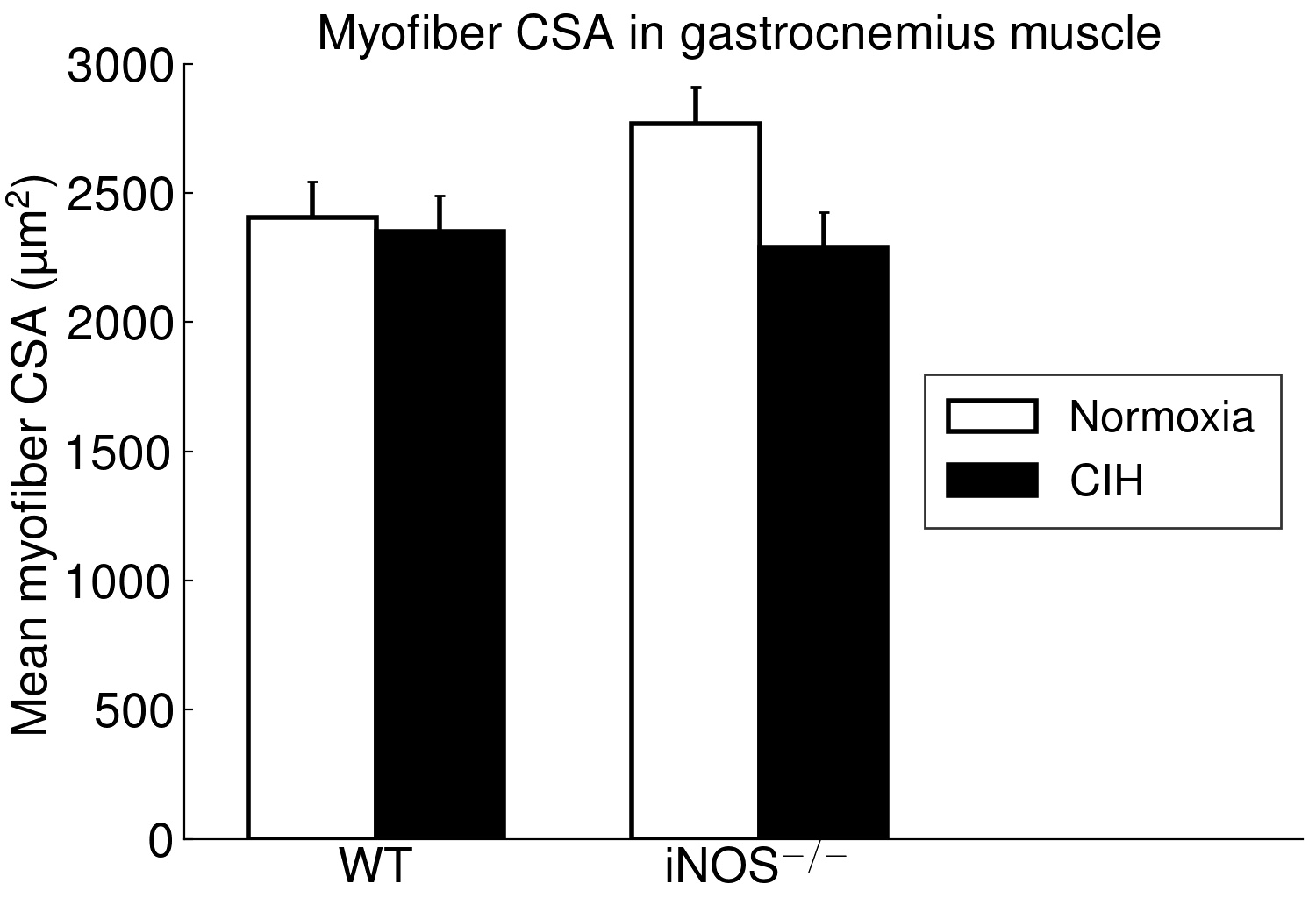
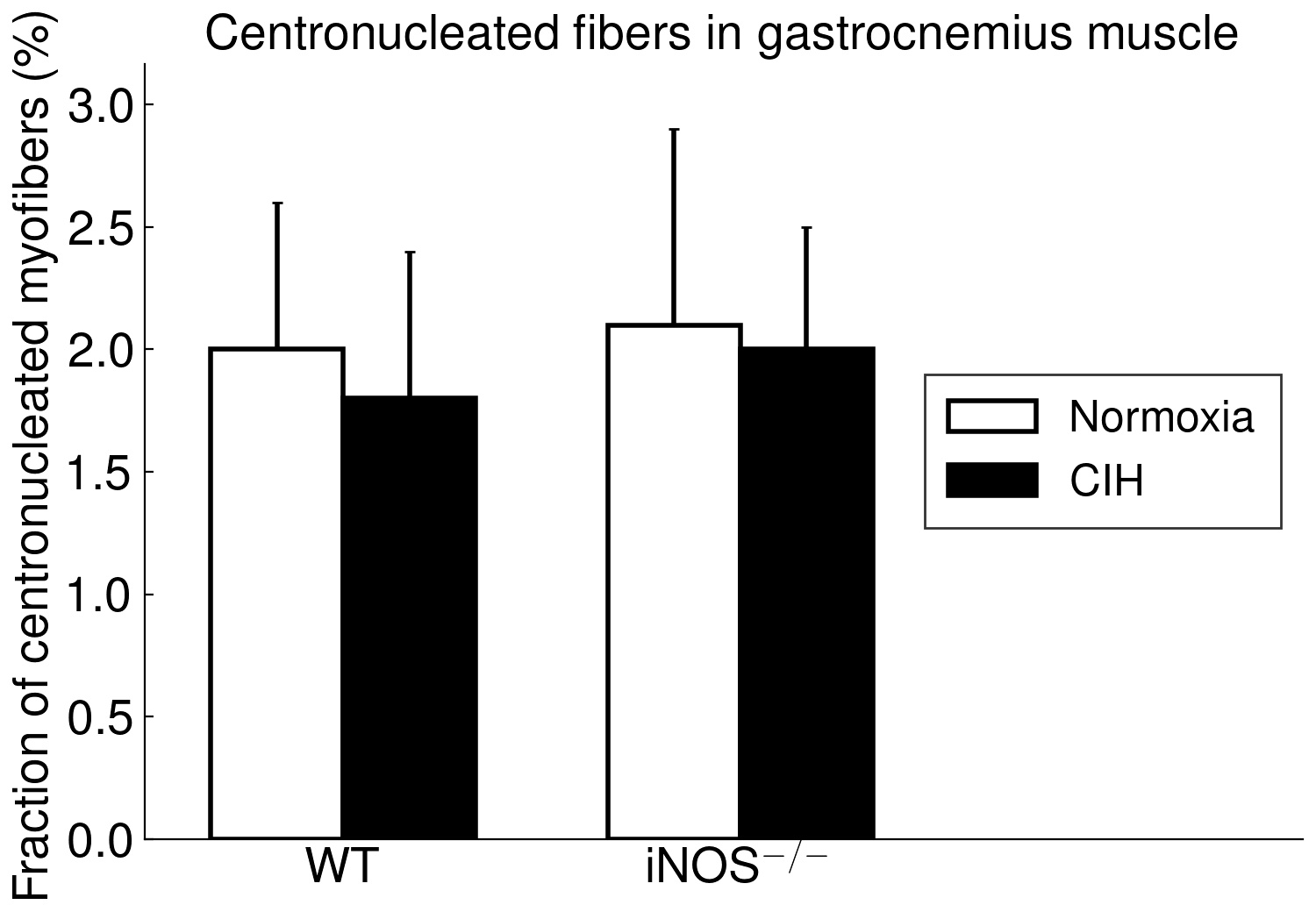
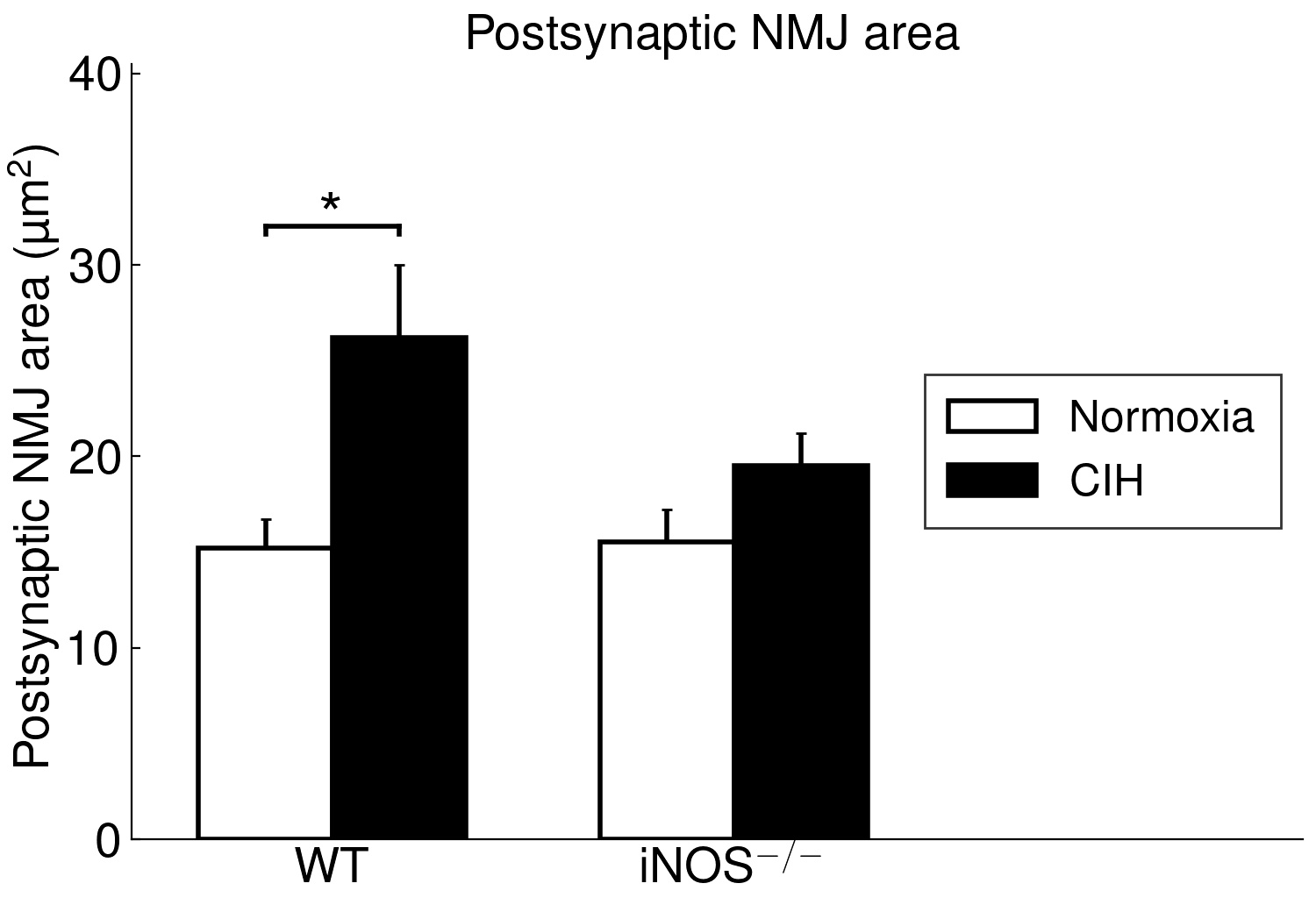
The present study furthermore included iNOS−/− mice into this analysis of CIH vs. NOX effects on skeletal muscle, in order to test the hypothesis that iNOS deficiency may at least in part protect against a pro-inflammatory/-oxidative effect through CIH, i.e. hypoxia-reoxygenation stress leading to ROS generation from various sources [36]. Contrary to expectation, our data demonstrate that, compared to WT, iNOS−/− by itself (i.e. under NOX conditions) also leads to highly significant postsynaptic NMJ area reduction and fragmentation in combination with mitochondrial damage and swelling, which surprisingly resemble and exceed those observed with CIH in WT mice. Notably, under the conditions of iNOS deficiency, CIH stress is able to further aggravate the damage at least in terms of a further reduction in postsynaptic NMJ area or length after normalization for fiber CSA or perimeter, respectively. The similarity between CIH (compared to NOX in WT) and iNOS−/− (compared to WT in NOX) was limited to NMJ and mitochondrial damage, while decreases in fiber CSA (including its correlation to NMJ) and centronucleation observed with CIH vs. NOX in WT were absent in iNOS−/− mice, i.e. they revealed no atrophy despite signs of denervation.
As another striking similarity, we found a > 10-fold increase in SOCS3 expression with CIH vs. NOX in WT as well as with iNOS−/− vs. WT at NOX in (pooled samples of) soleus muscle. Available evidence qualifies SOCS3 as a candidate to mechanistically link mitochondrial damage to NMJ deterioration: SOCS3 upregulation has been demonstrated as an early event after skeletal muscle denervation by sciatic nerve transection [37]. This obviously occurs in response to local inflammatory signals, especially via IL6 which by itself, i.e. without denervation, causes upregulation of SOCS3 and of E3-ligases (atrogin-1, MURF-1) together with fiber atrophy all of which is abrogated by IL6 inhibition. At the same time, SOCS3 overexpression has been shown to cause mitochondrial damage like swelling or disruption in tibialis anterior muscle, which is reminiscent of what was presently observed in soleus muscle but not in gastrocnemius muscle. SOCS3 overexpression was, furthermore, associated with inhibited expression of mitochondrial genes, which included Smtck and Slc25a3 [38], but may also comprise mtSOD, which was presently found to be downregulated. As an inhibitor of leptin and insulin signaling, increased muscular SOCS3 expression has been suggested as a major contributor to mitochondrial dysfunction, impaired fatty acid oxidation, as associated with aging, metabolic syndrome and inflammation [39–41]. These severe metabolic effects in combination with previous evidence that SOCS3 overexpression dilates the sarcoplasmatic reticulum, dislocates and inhibits calcineurine (colocalized with SOCS3) and reduces skeletal muscle energy expenditure, oxygen uptake and activity [38] may contribute to muscle fiber atrophy as observed in case of CIH. While we found no evidence for increased apoptosis signals, a SOCS3 upregulation appears to be associated with impaired regenerative stem cell function in elderly humans [39] and may potentially play a role in the increased centronucleation presently observed with CIH.
Moreover, the moderate decrease in mtSOD expression presently observed in soleus muscle with CIH and, to a lesser extent, with iNOS (NOX or CIH) might also play a role in fiber atrophy and mitochondrial deterioration: Sod1−/− mice, used as a murine model of neuromuscular impairment in age-related muscle atrophy (sarcopenia), exhibit reduction in myofiber CSA of type IIa fibers [42, 43]. Reduced CSA of type IIa fibers was, indeed, presently observed in association with the most marked mtSOD decrease (ca 50%), lower weight gain and alterations in mitochondrial ultrastructure and NMJ morphology. In addition, the observed shift in fiber metabolic phenotype, i.e. an increased type 1 at a decreased type 2 fiber fraction with both CIH (vs. WT) and iNOS−/− (vs. WT at NOX) might be attributed to decreased mtSOD expression, rather than to SOCS3 upregulation which decreases oxidative fiber characteristics [38]. Deficiency in mtSOD, representing impaired antioxidant defense, may also be involved in a remarkable number of age-related features which may originate from a loss of fast motoneurons followed by a reinnervation of slow motoneurons [44]. In the present study, fiber type ratio (type 1/type 2a) was significantly correlated with NMJ fragmentation and inversely correlated with NMJ size, pointing at a role of reinnervation in the fiber type shift.
Thus, our observations with CIH may display some analogies to age-related neuromuscular deterioration involving SOCS3 and mtSOD. They are in line with previous studies in other rodent CIH models, revealing downregulation of mtSOD/SOD2 via downregulation of HIF-2α [45] and clinical observation of lower plasma CuZnSOD/SOD1 in OSA patients [46].
Our CIH-based OSA mouse model is, however, at variance with biopsy studies in OSA patients, which revealed no changes in tibialis anterior muscle fiber size compared to controls [11] or showed even enlarged diameters of type 2a fibers in quadriceps femoris at unaltered fiber type composition [47]. One should, however, bear in mind, that the CIH mouse model does not mimick certain OSA-inherent factors like sways in intrathoracic pressure and blood pCO2 as well as ventilatory overshoots but at the same time involves more severe O2-desaturation without airway obstruction. Also the genetic background of mice may affect the degree of atrophy [48].
Even more important, our data provide first evidence for a strikingly differential effect of CIH-exposure between soleus and gastrocnemius i.e. (mixed) ’red’ and ‘white’ muscles, that has to be taken into account in translational studies. Indeed, in contrast to soleus muscle, gastrocnemius muscle revealed neither mitochondrial damage nor NMJ alterations (rather enlargement than shrinkage) with CIH. The differential exertion profiles between the postural soleus muscle (remaining recruited throughout during quiet standing) and the locomotor gastrocnemius muscle (providing fast forceful contractions) [49] may impact these muscle-specific findings in CIH and iNOS−/− mice and have likewise been implicated in massive muscle- (fiber-) specific differences in muscle aging or neurodegenerative disease [50–52].
However, our findings of compromised mitochondrial ultrastructure and gene expression in soleus muscle of CIH-mice may be in line with those in human palate muscle (a primary research focus within OSA pathophysiological), showing abnormal mitochondrial function and organization [53]. The observed close positive correlation of the fraction of damaged mitochondria to fragmented NMJ (or inverse correlation to NMJ area) reveals no clue for cause-effect relationship. As a first assumption, mitochondria-derived oxidative stress during CIH (hypoxia-reoxygenation stress) may compromise NMJ [54] acting in combination with other ROS sources like upregulated NOX2, as reported for the presently used CIH mouse model [55]. However, importantly, iNOS−/− at NOX largely mimicked the CIH effects, i.e. NMJ and mitochondrial damage together with SOCS3 up- and mtSOD downregulation and was despite the fact that iNOS mRNA expression in soleus or gastrocnemius muscles in WT was neither detectable with NOX nor with CIH exposure. Since, in contrast, WT mice revealed a hepatic iNOS expression, which was massively and significantly reduced (> 8-fold) with CIH compared to NOX, it is reasonable to assume that iNOS deficiency outside the skeletal muscle conveys both, the CIH and iNOS−/− effects. Thereby the NMJ damage, similarly observed with both these condition, strongly points towards an iNOS deficiency in peripheral nerves (i.e. in perikaryon of motoneurons or Schwann cells) as a cause of NMJ damage, though myeloid iNOS expression may also become muscle-protective [56]. Peripheral nerve injury may dramatically upregulate the low constitutive iNOS expression in Schwann cells, and iNOS deletion may result in smaller regenerating myelinated fibers and delayed reinnervation of muscle NMJ distal to the injury [57]. In fact, peripheral nerve dysfunction in patients suffering from OSA appears to be an early event [58], and denervation may precede muscular dysfunction, as suggested for human upper airway muscles [37, 59, 60] and supported by increased sarcolemmal N-CAM staining [15]. To date, no corresponding neuromuscular data exist for human locomotor muscle with OSA. However, they are needed to evaluate functional relevance of these alterations and to separate OSA-specific effects on NMJ, mitochondria, metabolism and related fiber dysfunction from the processes of aging and degenerative diseases [22, 61, 62]. Notably, the age of mice presently under test (four months) corresponded to early human adulthood (20–30 years) [63].
The conclusion that iNOS expression (outside skeletal muscle) may be neuro-protective and relevant for ‘red’ (aerobic) muscle function may be somewhat counterintuitive, as iNOS upregulation is resulting in a boost of NO, that is antimicrobial or antitumoral but also cytotoxic to normal tissue [64]. Furthermore it is causally implicated e.g. in insulin resistance and diabetes. Nonetheless, in humans a basal NO production rate (rendered mostly but not exclusively by nNOS and eNOS) is physiologically required (reviewed by [34]). There is evidence that NO may convey physiological oxidative signals [65] and a certain production by iNOS is required for neuroprotective antioxidative defense [66], e.g. through the ROS scavenging function of NO [64, 66]. Data on skeletal muscle tissue are scarce, however, it was reported that iNOS deficiency leads to mitochondrial damage in myocardial dysfunction (adriamycin-based mouse model). Interestingly, this effect was abrogated by overexpression of mtSOD [67, 68], which presently was found to be downregulated with both, CIH or iNOS deletion.
As a limitation, this study includes no functional data regarding NMJ and skeletal muscle to challenge the relevance of morphological alterations. Moreover, our mouse model involved a limited CIH exposition of 5 days per week, which may allow adaptive or protective effects of 2 normoxic days per week. Nonetheless a previous study showed, that the pathophysiological changes of the clinical OSA, such as arterial hypertension, are accurately reflected by the here used CIH mouse model [55].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}