Sampling and sequencing
A naturally dead L. relictus fledgling from Hongjian Nur (39°04' N, 109°53' E), Yulin, Shaanxi Province, was collected and identified by H. Xiao, and the specimen (voucher number YG01) was deposited in the animal specimens museum of the Shaanxi Institute of Zoology, Xi’an, Shaanxi Province, China. Our team is a wildlife protection agency under the Xi'an branch of the Chinese Academy of Sciences, cooperating and working with the authority department on Hongjian Nur for nearly 20 years, mainly devoted to the protection of the relict gull. To protect Larus relictus, this project has been approved and received permission from the Nature Reserve Authority of Hongjian Nur.
DNA was extracted from the muscle using the CTAB method, and the DNA concentration and quality were measured using a NanoDrop 2000 and a Qubit Fluorometer, respectively. Both Illumina HiSeq 4000 and PacBio RSII sequencing pipelines were used. For the Illumina pipeline, five short fragment paired-end libraries (three of 270 bp and two of 350 bp) were constructed using the standard Illumina pipeline. The details of library construction are as follows. The genomic DNA was broken randomly using the ultrasonic method, and target fragments were filtered. The small fragment sequencing library was constructed through the steps of end repair, addition of polyA and adaptor, selection of target-size fragments and PCR. The size and quality of the library were evaluated using an Agilent 2100 and qPCR. The Illumina HiSeq 4000 sequencer was used for sequencing, with PE=150. For the long fragment library in the PacBio pipeline, the details of library construction are as follows. The genomic DNA was sheared using g-TUBE, followed by DNA damage-repair and end-repair. The dumbbell-type adapters were ligated, and exonuclease digestion was performed. BluePippin was used to select segments to obtain the sequencing library. In addition, total RNA was extracted from the heart, liver, spleen, lung and kidney of L. relictus using TRIzol, and RNA concentrations were measured using NanoDrop 2000, Qubit 2.0 and Agilent 2100. The Illumina HiSeq 4000 platform was used for sequencing RNA data.
Genome assembly assessment
Whole-transcriptomic data from the liver and an equal mix of five tissue RNA samples were used to assist genome annotations. Raw reads were filtered to remove adapter sequences and low-quality data, with clean reads assembled using Trinity [26]. After filtering out low-quality and short-length PacBio reads, LoRDEC [27] software was used for error correction of PacBio data employing HiSeq data. The HiSeq data were preliminarily assembled by Platanus [28] software. Using dbg2olc [29] software, mixed assembly was carried out by using the data after error correction and the preliminary assembly results of HiSeq data. Pilon [30] software was used to correct the assembly results using HiSeq data. To assess the completeness of the L. relictus genome assembly, we used two methods, with the first remapping the Illumina paired-end reads to the assembled genome and the second employing CEGMA v.2.5 [31] and BUSCO v3.0.2 databases.
Genome Annotation
Three different strategies were used to predict gene structures, namely, ab initio-based, homologue-based and RNA-seq-based methods. EVM v1.1.1 [32] software was used to integrate the predicted genes and generate a consensus gene set. Then, GENSCAN [33], Augustus v2.4 [34], GlimmerHMM v3.0.4 [35], GeneID v1.4 [36] and SNAP (version2006-07-28) [37] were first used to perform the ab initio prediction. For homologue prediction, GeMoMa v1.3.1 [38] was used, primarily employing five species as references, i.e., G. gallus, Meleagris gallopavo, Taeniopygia guttata, Ficedula albicollis and Parus major. Third, HISAT v2.0.4 and StringTie v1.2.3 [39] were used for assembly based on RNA-seq reference data, and TransDecoder v2.0 [40] and GeneMarkS-T v5.1 [41] were applied to predict genes. PASA v2.0.2 [42] was used to predict unigene sequences assembled based on the whole transcriptome data without references. Finally, EVMv1.1.1 [32] was used to integrate the prediction results obtained by the above three methods, and PASA v2.0.2 [42] was used to predict alternative splice variants.
Software including LTR-FINDERv1.05 [43], MITE-Hunter [44], RepeatScout v1.05 [45] and PILER-DF v2.4 [46] was used for prediction of repetitive sequences in the L. relictus genome. A combination of structure-based and de novo strategies was used to construct repeat databases and then merged with Repbase [47] to form a final database. RepeatMasker v4.0.6 was used to identify repeat sequences with this final repeat database [48].
Using the Rfam [49] and miRbase [50] databases as references, rRNA and microRNA were identified by Infernal 1.1 [51]. The tRNA was predicted using tRNAscan-SEv1.3.1 [52]. After screening the true loci of the genome, GenBlastA v1.0.4 [53] was used to blast to search homologous gene sequences. Pseudogenes were then identified via GeneWise v2.4.1 [54] with premature stop codons and frame shifts.
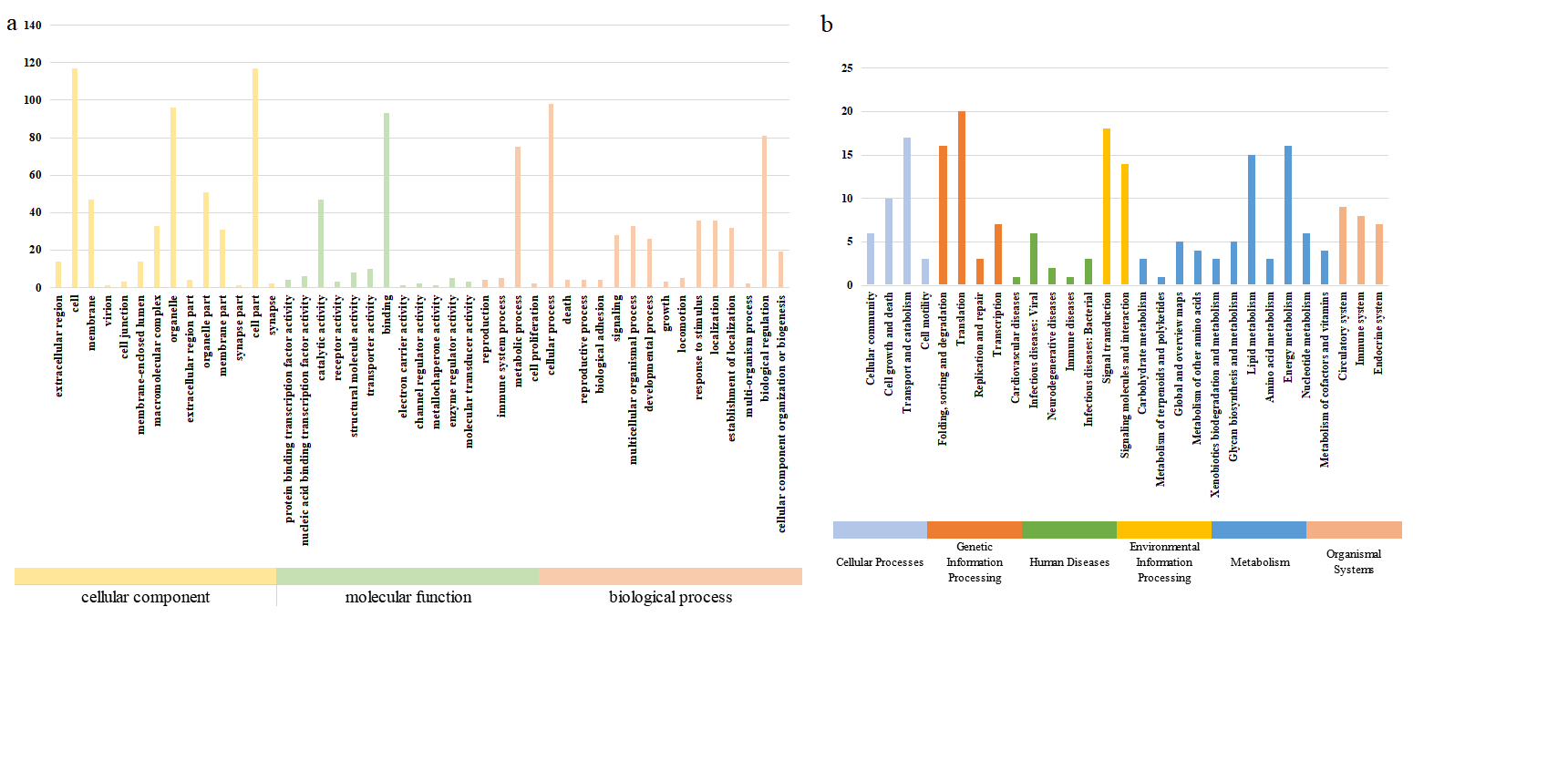
To assign gene functions in the L. relictus genome, we aligned the genes to five functional databases using BLASTv2.2.3 [55] (E-value = 1e-5). The databases included GO (Gene Ontology) [56], KEGG (Kyoto Encyclopedia of Genes and Genomes) [57], KOG (Cluster of Orthologous Groups for eukaryotic complete genomes) [58], TrEMBL (Translated EMBL-Bank) [59] and NR (NCBI non-redundant amino acid sequences) [60].
Phylogeny and positively selected genes
We used the whole-genome sequence of L. relictus and seven published whole-genome sequences of three Charadriiformes species (L. relictus, P. pugnax and C. vociferus), three Pelecaniformes (Phaethon lepturus, Nipponia nippon and Egretta garzetta), one Gruiformes (Mesitornis unicolor) and one Procellariiformes (Fulmarus glacialis). OrthoMCL was used to cluster gene families [61]. A total of 7,339 single-copy orthologues were identified, with protein sequences used for constructing phylogenetic trees. The protein sequences were aligned using MUSCLE [62] and then concatenated into a combined dataset. We then constructed the phylogenetic tree using the ML (maximum likelihood) algorithm with the JTT amino acid substitution model implemented in PhyML [63].
Based on the results of phylogenetic trees, divergence time was estimated using the MCMCTree program in PAML [64]. Divergence times and ages of fossil records were derived from TimeTree (http://www.timetree.org/) and applied as the time control. In addition, the CodeML program in PAML [65] included single-copy genes to detect positively selected genes in the L. relictus genome. Among them, the branch model (model=2, NSsites=0) was used to calculate ω0 (dn/ds) of the foreground clade and obtained the average ω1 of other clades. Then, model=0 and NSsites=0 were used to evaluate the ω2 of the whole tree, and those genes with ω0>ω2 were selected.
{kind=link}
{kind=link}