Lung microbiome description, gene prediction and species annotation
Of the 54 sputum samples, 38 were collected from the first acute exacerbation (period A). There were 12 stable period sputum samples (period B) from 6 patients (7 sputum samples were collected in the second month of the stable phase, 4 sputum samples were collected in the 6th month of the stable phase, and 1 sputum was collected in the 12th month of the stable phase). There were 4 sputum samples (period C) from 4 patients during the second acute exacerbation (2 samples were collected on the first day of admission and 2 samples were collected on the third day of admission) (Table 1).
We obtained 310,383 ORFs (Open Reading Frames), with a total length of 173.46 Mbp, GC content of 44.78%, from which 115,179 were assigned to genes. In the ORFs that can be annotated to the NR database (Version: 2018.01) from NCBI, 94.28% were assigned at the kingdom level, 91.89% at the phylum level, 90.35% at the class level, 87.81% at the order level, 85.51% at the family level, 82.71% at the genus level, and 47.85% at the species level(Additional file: Figure S1). Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria were the top 4 phylums. Streptococcus, Neisseria, Haemophilus and Prevotella were top 4 genus in the cohort, in which many members in these four genus were natural inhabitants in the human up respiratory tract(Figure 1)[18].
Difference of microbiome between stable and acute exacerbation stage of COPD
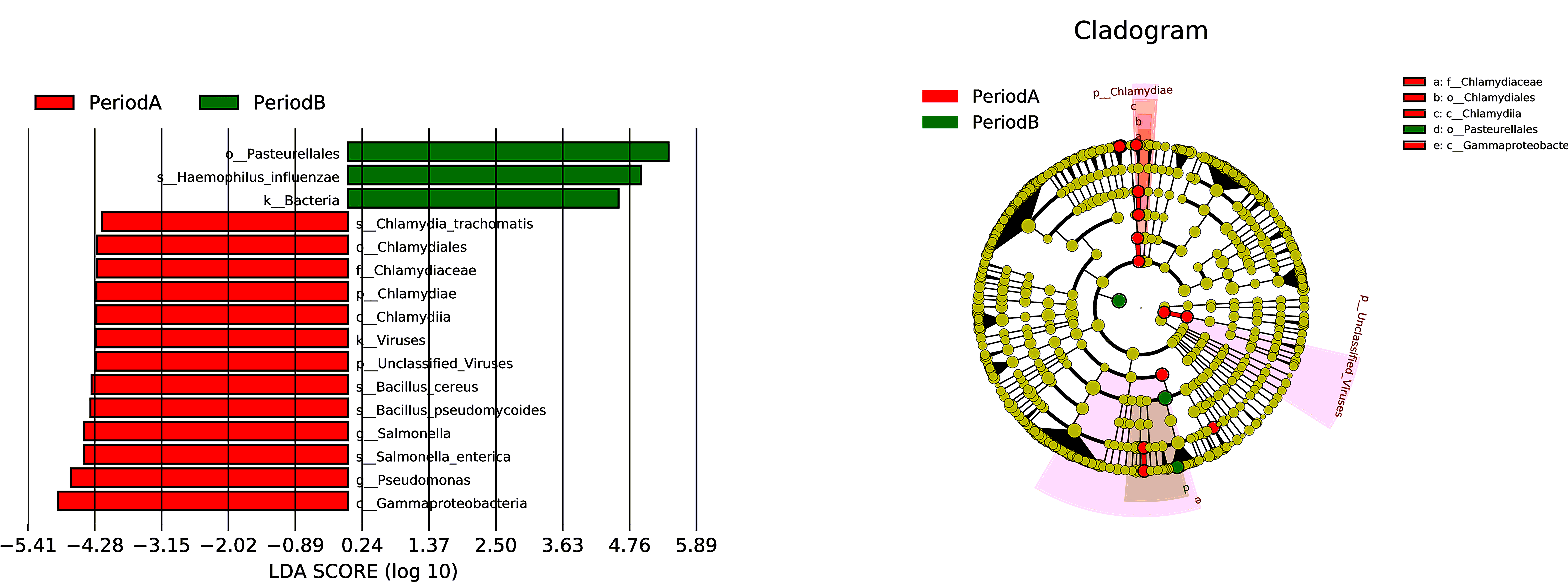
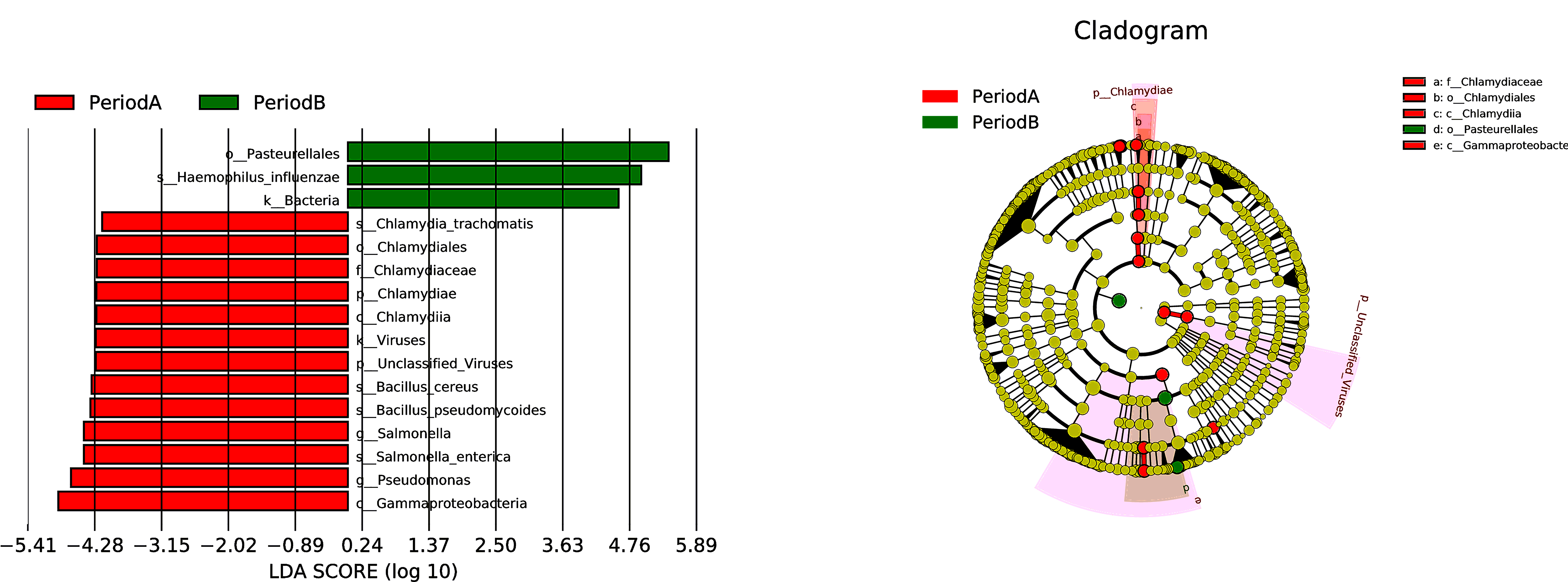
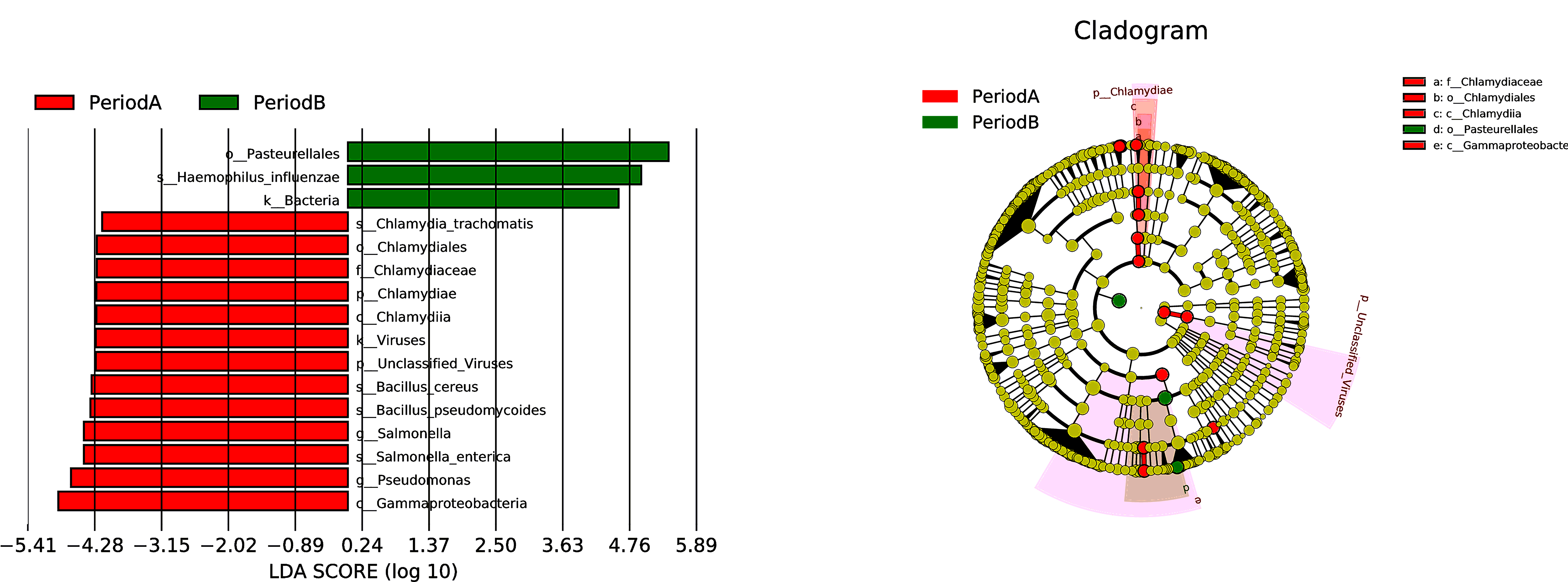
Our results show that the sputum microbiome composition of patients with acute exacerbation of COPD is different from that of patients at stable stage. At the phylum level, the relative abundances of Firmicutes and Proteobacteria in stable patients were 15.80% and 23.82%, respectively, while those in patients with acute exacerbation were 13.50% and 13.05% respectively, with no statistical difference between the two stages. At the genus level, the relative abundance of Haemophilus in stable patients was 10.32%, while that in patients with AECOPD was 2.71% (Figure 2, Additional file: Table S1, Additional file: Table S2), with no statistical difference between the two. The results of LEfSe analysis showed that in period A group, Salmonella, Salmonella enterica, Pseudomonas and Proteobacteria achieved high abundance. While in period B, Haemophilus influenzae, Pasteurellales were more abundant (Figure S2). In addition, period A cohort showed higher α diversity than period B group (Figure 3).
Analysis of gene function and resistance genes
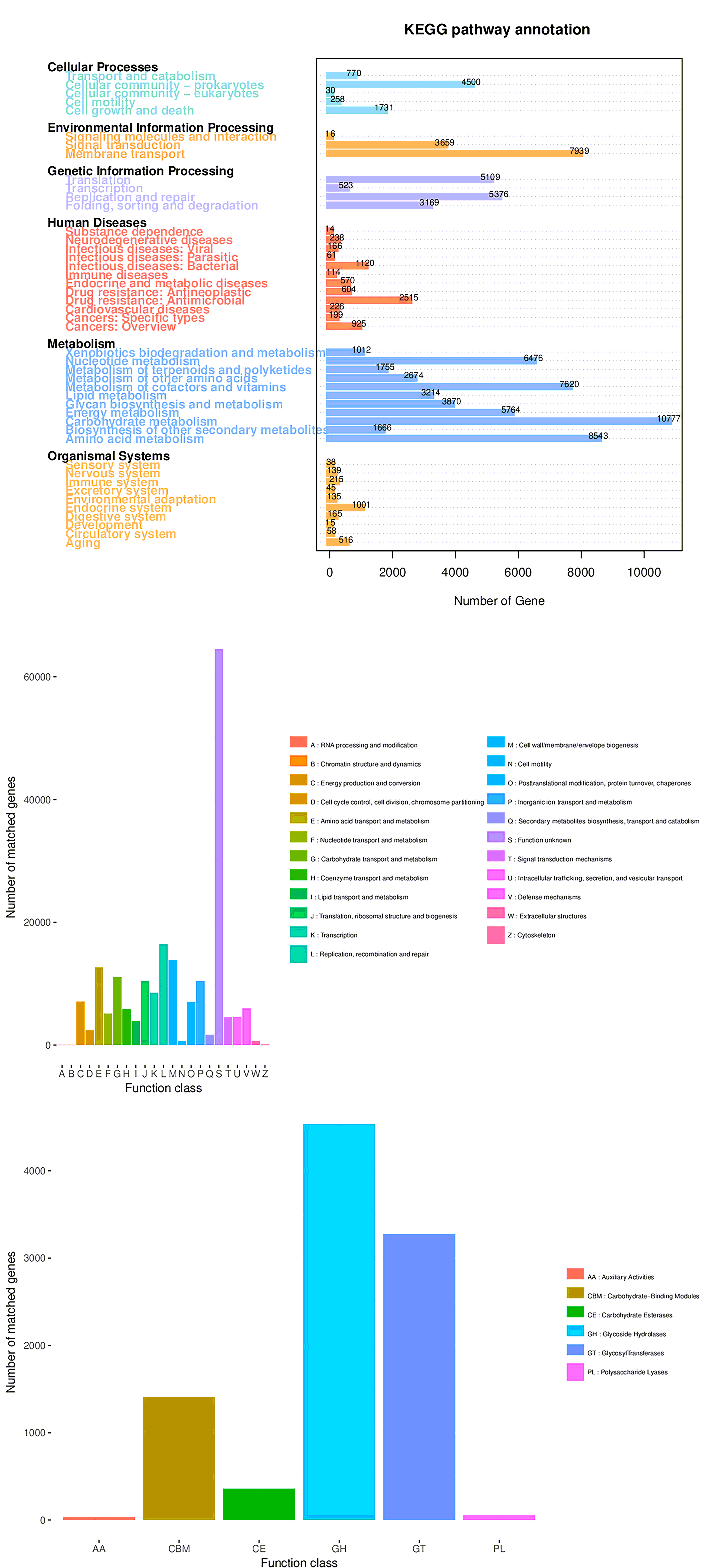
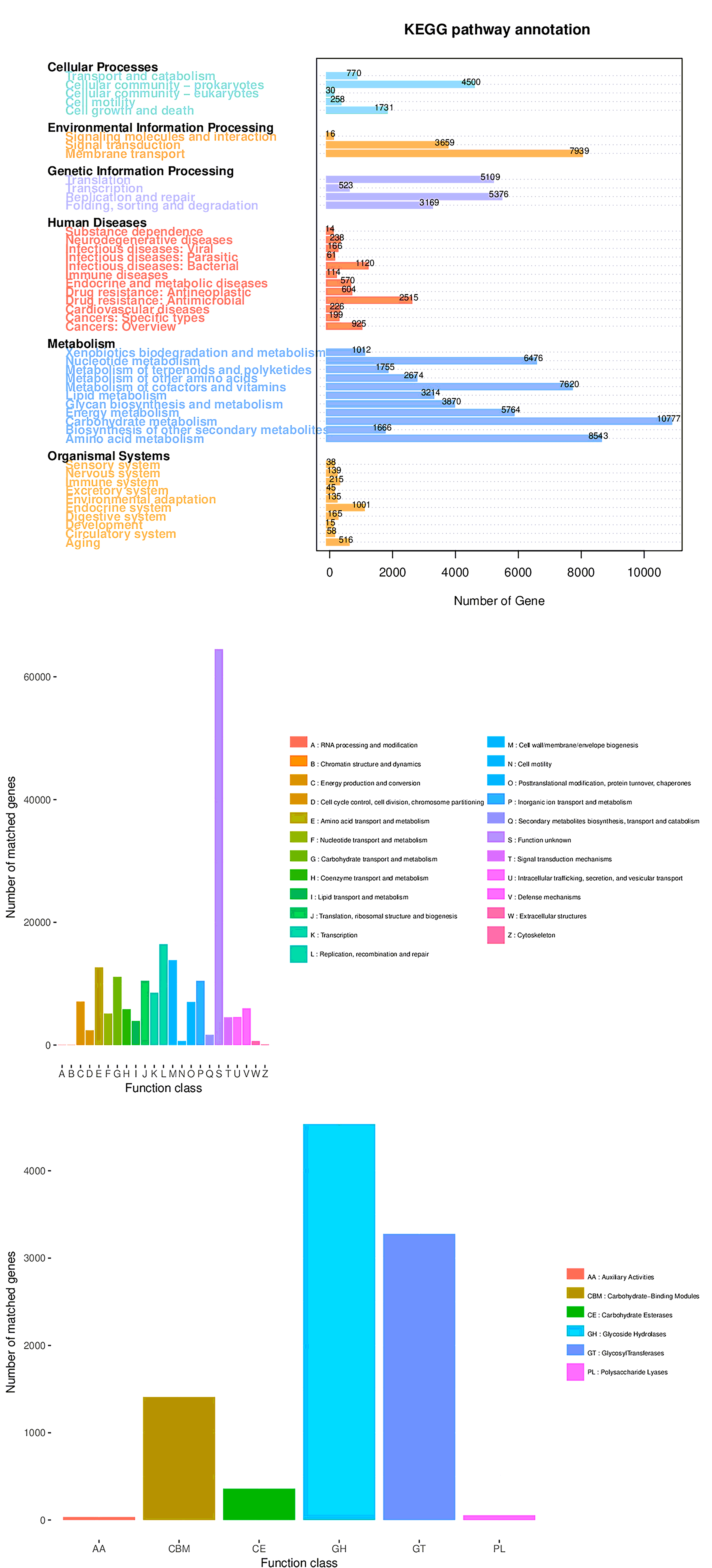
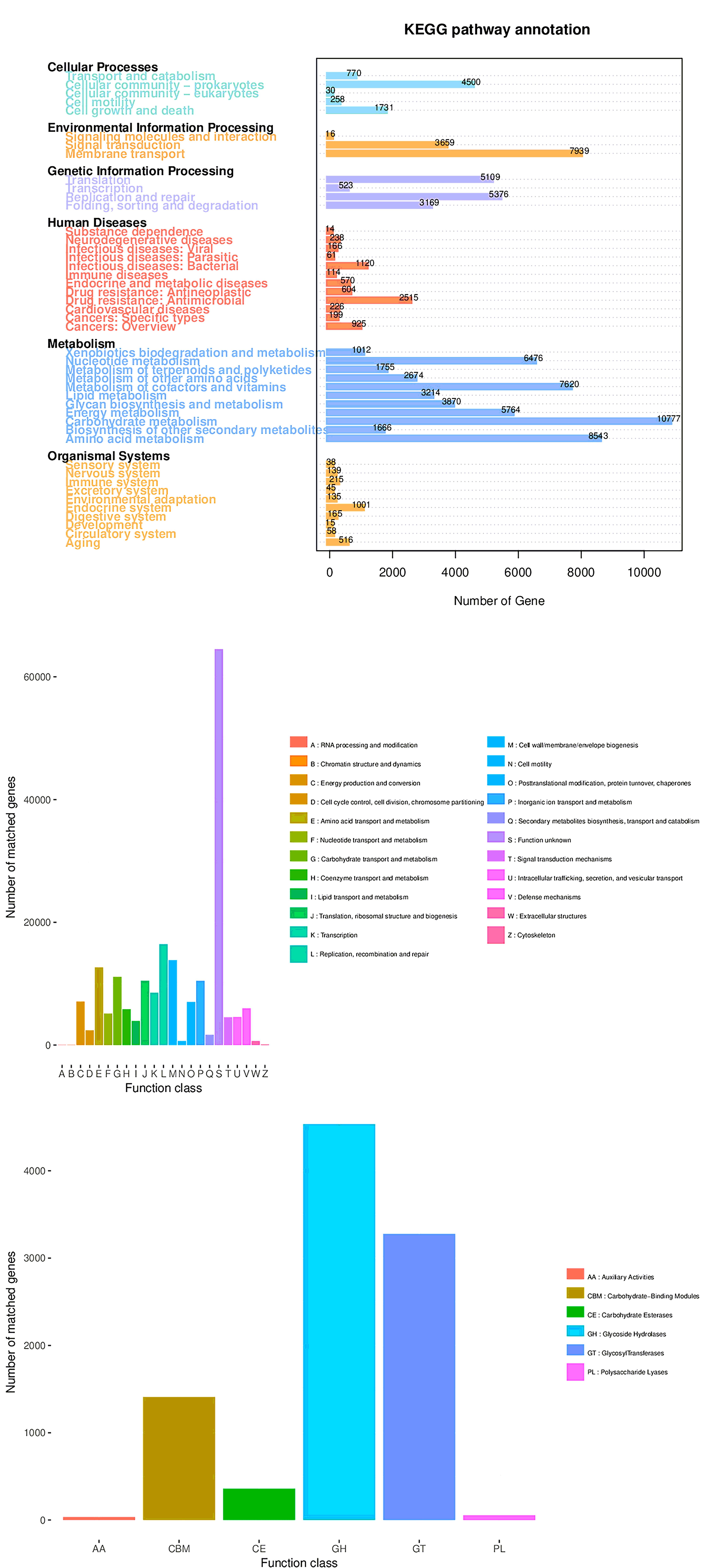
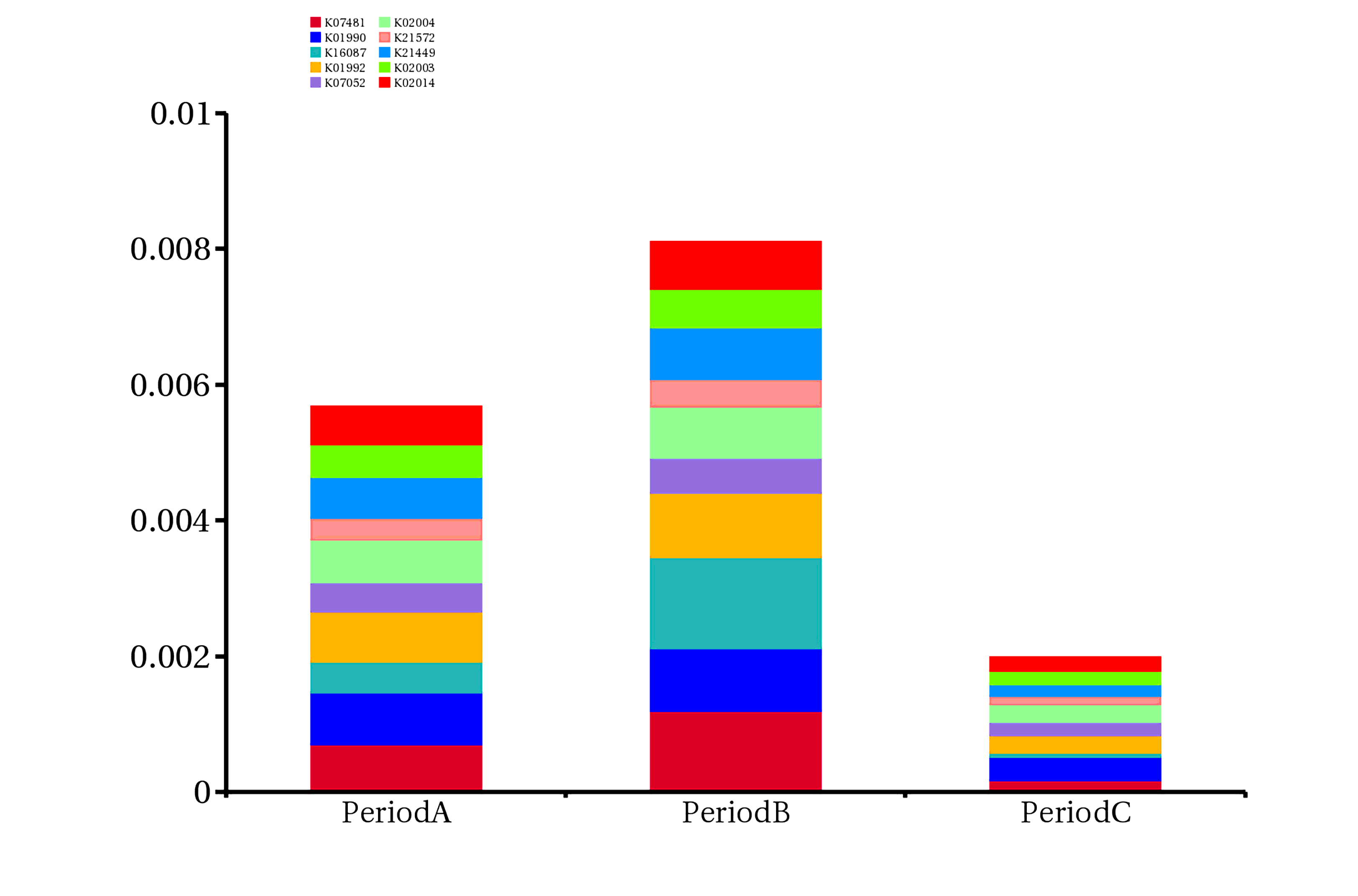
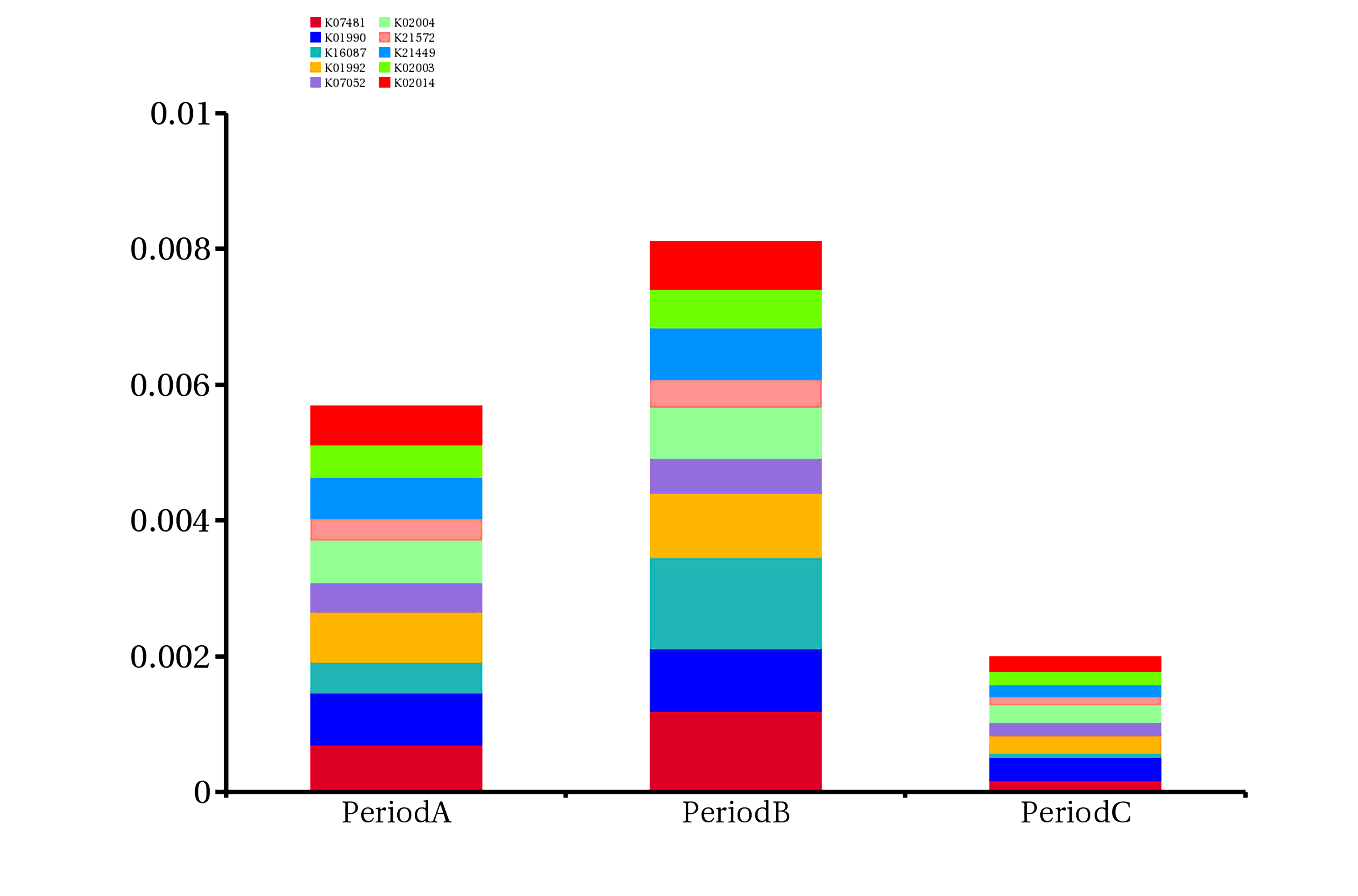
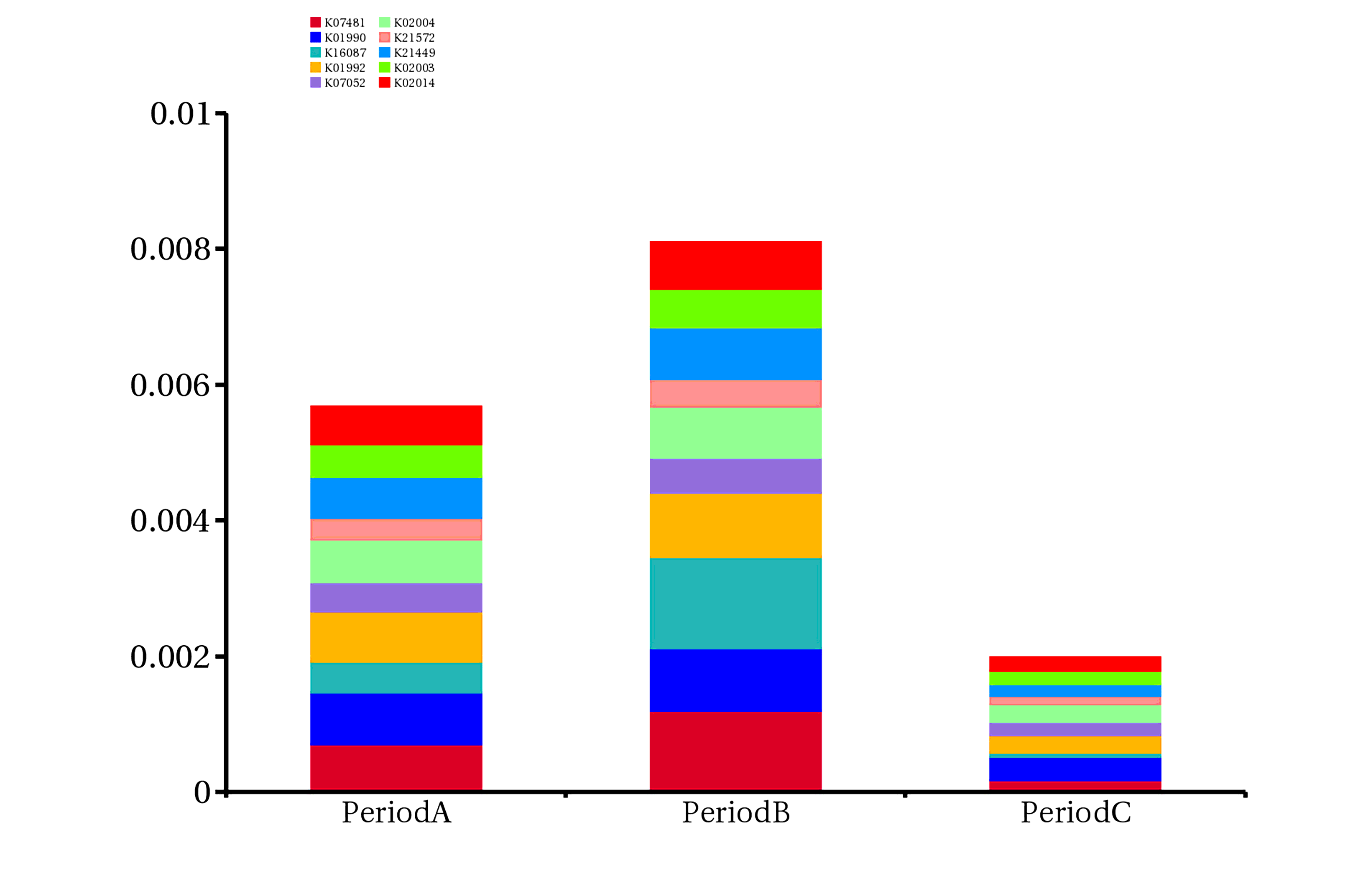
In order to further evaluate the function of genes, genes obtained by sequencing were annotated with the following functional database (KEGG, eggnog and CAZy database) by DIAMOND software. There are 201,130 (64.80%) genes that can match the KEGG database, of which 108,824 (35.06%) can match 4,562 KEGG ortholog group (KO) ,193014 (62.19%) genes can be assigned to the eggNOG database,8984 (2.89%) genes can be matched the CAZy database. (Figure S1). We found that the relative abundance of genes annotated to K07481 and K16087 in period B subgroup was higher than that in period A or C subgroup. The relative abundance of the former in period B subgroup was 0.119%, and that in period A and C subgroup was 0.07% and 0.017%, respectively. The relative abundance of the latter in period B subgroup was 0.135%, while that in period A and C subgroups was 0.046% and 0.006%, respectively (Figure S3, Additional file: Table S3). The results of resistance gene annotation showed that 275 genes could be found in the CARD database, including 198 kinds of ARO (the Antibiotic Resistance Ontology) Compared with patients with stable COPD, Proteobacteria harbored more ARO genes (44%) in patients with acute exacerbation, suggesting that drug resistance of Proteobacteria may play an important role in the process of acute exacerbation of COPD. After assigning ARO to species, in subgroup period A, 44% of ARO belonged to Proteobacteria, 13% to Firmicutes, 4% to Actinobacteria, and 4% to Bacteroidetes. In period B subgroup, 44% of ARO belonged to Proteobacteria, 13% to Firmicutes, 3% to Actinobacteria, and 3% to Bacteroidetes (Fig. 4).
Metabolic pathway analysis
In this study, there was no difference in the number of genes in different metabolic pathways between period A and period B groups. However, it was found that the relative abundance of genes annotated to the metabolic pathway of purine metabolism (ko00230), ATP binding- cassette (ABC) transporters (ko02010), two-component signal transduction system (ko02020), and ribosome (ko03010) function, the homologous recombination function (ko03440) are higher in period B group than that in period A group (figure 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}