The purpose of this research was to determine Aspergillus fumigatus conidial viability and its biofilm formation upon treatment with triclosan and amphotericin-B loaded liposomes. A. fumigatus was treated with the antimicrobials, triclosan and liposomal amphotericin-B (L-AMB), in single and combined supplementation. To quantify the cells’ viability upon treatments, resazurin-based viability assay was performed. Confocal laser scanning microscopy was done by applying FUN-1 stain to screen the role of the agents on extracellular polymeric substances. Total A. fumigatus biomass upon treatments was estimated by using crystal violet-based assay. To study the agents’ effect on the conidial viability, flow cytometry analysis was performed. Expression levels of A. fumigatus genes encoding cell wall proteins, α-(1,3)-glucans and galactosaminogalactan were analysed by real-time polymerase chain reaction assay. A synergistic interaction occurred between triclosan and L-AMB when they were added sequentially (triclosan+L-AMB) at their sub-minimum inhibitory concentrations, the triclosan and L-AMB MICs were dropped to 0.6 and 0.2 mg/L, respectively, from 2 and 1 mg/L. Besides, L-AMB and triclosan contributed to the down-regulation of α-(1,3)-glucan and galactosaminogalactan in A. fumigatus conidia and resulted in less conidia aggregation and mycelia adhesion to the biotic/abiotic surfaces; A. fumigatus conidia -became hydrophilic upon treatment, as a result of rodlet layer being masked by a hydrophilic layer or modified by the ionic strength of the rodlet layer. In A. fumigatus, the potential mechanisms of action for L-AMB might be through killing the cells and for triclosan through interrupting the cells’ development as a consequence of quorum quenching.
Research Article
Antifungal Effect of Triclosan On Aspergillus Fumigatus: Quorum Quenching Role As A Single Agent And Synergy With Liposomal Amphotericin-B
https://doi.org/10.21203/rs.3.rs-1039223/v1
This work is licensed under a CC BY 4.0 License
You are reading this latest preprint version
Conidia
biofilm
combination treatment
flow cytometry
quorum quenching
Biofilm formation is implicated in the pathogenesis of localized as well as invasive Aspergillus fumigatus (A. fumigatus) diseases (Beauvais et al. 2007; Mowat et al. 2007; Chandrasekar and Manavathu 2008). Aspergilloma, a localized infection, and invasive pulmonary aspergillosis (IPA) have been shown to form biofilms containing highly agglutinated hyphae of A. fumigatus encased in extracellular polymeric substances (EPS) (Mowat et al. 2008; Loussert et al. 2010). Amphotericin B (AMB) is an antifungal agent and as a polyene antibiotic, it binds to ergosterol in the fungal cell membrane and through a series of steps destroys the cells. However, as biofilms are highly resistant to antimicrobials, A. fumigatus colonies encased in EPS are more resilient to AMB (Beauvais et al. 2007). It has been demonstrated that during early to intermediate phases of biofilm development, EPS are produced close to the fungi maturity stage (Ramage et al. 2011). Microbial cells facilitate, through EPS production, the attachment of other pathogens by providing more diverse adhesion sites on biotic and abiotic surfaces. EPS holds the biofilm together, leading to an irreversible cell attachment to the surfaces (Flemming and Wingender 2010). Thus, EPS formation and degradation can be used as a marker for the antifungal activity of an agent.
As a major constituent in A. fumigatus biofilm EPS, Galactosaminogalactan (GAGs) plays a critical role in maintenance of the EPS (Loussert et al. 2010). Activity of sph3, which encodes a protein belonging to the spherulin 4 family is required for the synthesis of functional GAG (Bamford et al. 2015). Strains deficient in GAG fail to produce EPS and are unable to form adherent biofilms on plastics or host cells in vitro (Gravelat et al. 2013). GAG-mediated adherence is largely a consequence of charge interactions between the polycationic polysaccharides of the fungus cell wall and negatively charged surfaces (Lee and Sheppard 2016). Cell wall α-(1,3)-glucans play an essential role in conidia (asexual spores) aggregation (Fontaine et al. 2010; Henry et al. 2011). In particular, three α(1–3)-glucan synthase genes, ags1, ags2, and ags3, have been found to be responsible for cell wall α(1–3)-glucan biosynthesis. Among these genes, only ags3 is involved in virulence (Beauvais et al. 2005; Maubon et al. 2006). Taken together, α-(1,3)-glucan and GAG down-regulation might result in less conidia aggregation and mycelia attachment to the biotic/abiotic surfaces, respectively.
Quorum sensing (QS) signals contribute directly to microbes’ pathogenesis due to the production of virulence determinants such as toxins, proteases, and immune-evasive factors (Scoffone et al. 2019). Quorum quenchers as biostatic agents affect QS pathways and hence biofilm formation. Biostatic agents are increasingly favoured over microbicidal agents as they do not appear to trigger resistance in microbes (Martínez et al. 2019 and Lu et al. 2021). Targeting components of microbes that are responsible for pathogenesis, makes quorum quenchers better candidates compared to the agents with growth-inhibitory effects.
Triclosan (C₁₂H₇Cl₃O₂) is an antiseptic diphenyl ether derivative used in toothpaste, mouthwash, surgical soaps, and cosmetics (Weatherly and Gosse 2017). It disrupts biofilm formation in some bacteria, yeasts, and dermatophytes through blocking the biosynthesis of amino acids and fatty acids (Hoang and Schweizer 1999 and Jones et al. 2000). Features such as nuclear chromatin condensation, substantial intracellular vacuolation and mitochondrial swelling were shown by triclosan-treated Cryptococcus neoformans (C. neoformans) suggesting that triclosan induced apoptosis-like cell death (Movahed et al. 2016). However, the efficacy of triclosan against filamentous fungi has not been studied so far.
Synergism has been identified between triclosan and two principal drugs, AMB and fluconazole against C. neoformans (Movahed et al. 2016). The study applied microdilution checkerboard assay and viewed cell morphology under transmission electron microscope. In Tobudic et al. (2010) synergism was demonstrated by the combination of AMB/Posaconazole with improved effectiveness against C. albicans biofilms. Mesquita et al. (2013) used flow cytometry to investigate the effect of disinfection therapy on the survival, development, and metabolic activity of gamma-radiated fungal spores (Penicillium chrysogenum, Aspergillus nidulans, and Aspergillus niger).
Resistance of A. fumigatus towards AMB, and the side effects of single use triclosan as a synthetic antimicrobial agent on the environment and human health are the concerns which can be overcome through combination therapy.
In this study we used liposomal AMB (L-AMB), which compared to the conventional AMB deoxycholate, has the same antifungal activity following its incorporation into a liposome bilayer, while its toxicity against human organs is significantly lower (Stone et al. 2016). We also investigated the potential quorum quenching role of triclosan against A. fumigatus and compared it to the efficacy of L-AMB. Furthermore, we studied the combination of triclosan and L-AMB against A. fumigatus viability.
Persister cells (also called drug-tolerant cells), which include approximately ≤ 1% of the overall biofilm population, are considered as a possible reason for drug resistance. Persister cells neither grow nor die in the presence of microbicidal agents (Keren et al. 2004; Lewis 2007). Triggering persister cells by an antimicrobial agent, and hence making them vulnerable against another agent, is considered as a strategy for a combination treatment. In this context, we investigated the synergy between triclosan and L-AMB through their effect on triclosan triggered persister cells.
The fungus and its maintenance
A. fumigatus ATCC46645, was obtained from the Culture Collection of the University of Westminster, London, UK. Stock cultures of A. fumigatus maintained on potato dextrose agar (PDA) (Merck, Dorset, UK,) medium were propagated in potato dextrose broth (PDB) (Fisher Scientific, Loughborough, UK) or RPMI-1640 (Merck, Dorset, UK).
Culture and treatment of A. fumigatus for resazurin-based viability assessment
Minimum inhibitory concentration (MIC) is defined as the lowest concentration of an antimicrobial that will inhibit the visible growth of a microorganism after overnight incubation (Andrews 2001). Different doses of the antimicrobial agents, triclosan (Sigma-Aldrich, Dorset, UK) and L-AMB (Thermo Fisher Scientific, Leicestershire, UK) were applied to A. fumigatus at t0 and after 24 (represents early biofilm structure) and 48 (represents mature biofilm structure) hrs. To measure fungus viability, resazurin dye solution (Merck, Dorset, UK) was added to the culture (10% v/v) in 96-well plates. The plates were then incubated at 37°C on a shaker at 50 rpm for 50 min and the absorbance was measured spectrophotometrically at a wavelength of 590 nm. Single and combination treatments of A. fumigatus with antimicrobial agents, triclosan and L-AMB, were performed. For that, conidia and sessile (biofilm) cell viabilities were measured by In vitro toxicology assay kit (Merck, Dorset, UK) in 96-well tissue culture plates (Corning Inc., Corning, NY).
Treatment of A. fumigatus with triclosan and L-AMB
For single treatment, in a 96-well microplate, RPMI-1640 medium (200 µL) containing the agent’s effective dose and inoculum (106 conidia/mL) was added per well. After incubation at 37°C for 24 h, the cells’ metabolic activity was determined through resazurin assay.
Triclosan-L-AMB interactions were evaluated using the checkerboard assay. Checkerboards were prepared by using serial dilutions of L-AMB and triclosan. Triclosan and L-AMB dilutions were prepared as recommended in the EUCAST (European Committee on Antimicrobial Susceptibility Testing) protocol to give final drug concentrations of 0.3, 0.6 and 1.2 mg/L and 0.1, 0.2 and 0.4 mg/L for triclosan and L-AMB, respectively, in 100 µL of double-strength RPMI medium containing 106 conidia/mL fresh spore suspension. Subsequently, 100 µL of RPMI medium containing fresh spore suspension (106 conidia/mL) were added to the wells, resulting in concentrations of 0.15, 0.3 and 0.6 mg/L and 0.05, 0.1 and 0.2 mg/L for triclosan and L-AMB, respectively. For both simultaneous and continuous combination treatment strategies, viability was determined by using resazurin assay after 48 h treatment at 37°C. The OD at 570 nm wavelength was determined with a spectrophotometer (Jenway 6300 visible; Camlab Limited, UK). Fractional inhibitory concentration index (FICI) was calculated as follow:
(FICI=(MICAcomA,B÷MICagentA)+(MICBcomA,B÷MICagentB)) Equation 1
According to the above equation, FICI ≤ 0.5 indicates synergy, FICI > 4 indicates antagonism whereas 0.5 < FICI < 4 suggests no interaction. To study the synergistic assay, Compusyn software (ComboSyn, Inc.) was used. The resulting CI theorem of Chou-Talalay offers quantitative definition for additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1) in drug combinations (Chou 2010).
Evaluation of the EPS and biofilm depth by using confocal laser scanning microscopy
Leica SP2 LSCM (CLSM, Carl Zeiss, Jena, Germany) was used to examine the fluorescent filamentous biomass and hence the biofilm and its thickness. A. fumigatus biofilm formation on glass slides after 48 h of incubation at 37°C were analysed using FUN-1-based confocal laser scanning microscopy (CLSM). Microscopic visualization and image acquisition of biofilms were conducted using an upright scanning Leica confocal microscope equipped with an argon/krypton laser and detectors, and filter sets for monitoring of green (excitation 480 nm, emission 517 nm) and red (excitation 633 nm, emission 676 nm). The biofilms on the surfaces were washed three times in sterile PBS and stained using a fluorescent stain, FUN-1 (Molecular Probes, Life Tech) prepared according to the manufacturer’s instructions. For biofilm visualization, FUN-1 (1 µL) from a 10 mM stock was mixed in 1 mL of PBS. Three drops of the mixture were added on the top of the biofilm, which was then mounted on a coverslip. The slides were incubated for 45 min at 37°C in the dark. The biofilm, formed as explained above, was washed again with PBS and mounted on a slide. An excitation wavelength of 488 nm using an argon ion laser at a magnification of 200x was used to examine the biofilms. Horizontal (xy) view of reconstructed 3-dimensional images of FUN1-stained biofilms was applied to capture green fluorescence.
Thickness of the biofilm was observed in the side view of the reconstruction. Sections on the xy plane were taken at 1 µm intervals along the z-axis of the sections taken parallel to the x-y plane to determine the depth of the biofilms. The CLSM analysis of the biofilms was used to measure and to compare the “means” of the thickness of the triclosan-treated and the combination-treated test groups. Three-dimensional images were assembled using Leica Confocal Software.
Biofilm quantification by crystal violet assay
The effect of the agents (triclosan and L-AMB at their MICs) on the mature biofilm (36 h of incubation) was estimated using the crystal violet (CV) assay. The assay stains both live and dead cells as well as some components present in the biofilm matrix, thus it is well suited to quantify total biofilm biomass. To determine the ability to form biofilms, an inoculum (106 conidia/mL) was added to 200 µL PDB medium in a 96-well polystyrene microtiter plate, which was incubated, without agitation, at 37°C for 36 h. Subsequently, the medium was aspirated and non-adherent cells removed by thoroughly washing the formed biofilm three times with PBS. After fixation with glutaraldehyde (70% in H2O; Sigma-Aldrich, UK) for 20 min and air-drying, the biofilms were stained with 200 µL, 0.5% w/v CV (Merck, Dorset, UK) for 15 min, followed by rinsing with sterile PBS, and de-staining with 96% v/v ethanol (VWR, Brooklyn, NY). The absorbance of the CV- stained biofilm in the treated and untreated control groups was measured at 590 nm using a spectrophotometer. Percentage biofilm biomass in the treated samples was calculated using the following equation:

where A590 is the absorbance of the CV-stained biofilm matrix at 590 nm.
Viability assay of the conidia by using flow cytometry
propidium iodide (PI) is a positively charged, membrane-impermeable fluorochrome that can only pass through the membranes of stressed, injured, or dead cells. It has been already shown that flow cytometry can be applied on conidia cells to assay their viabilities (Balajee and Marr 2002; Mesquita et al. 2013; Vanhauteghem et al. 2017).
To assess conidia viability, protocol defined by Mesquita et al. 2013 was adopted and filtered sterile water was used instead of culture media. Flow cytometry tubes (3.5 mL) using control tubes and experimental tubes were prepared (1 mL of deionised water + 1 mL of the conidia suspension). Subsequently, 100 µL of each treated and untreated control group was added to a flow cytometry tube with 50 µL of PI (Abcam, Cambridge, UK) stock solution (1 mg/mL), and 1.85 mL deionised water. After 10 min of staining with PI, tubes were then mixed and analysed using the cytometer (NovoCyte Benchtop, ACEA Biosciences, UK). PI is excited at 488-523 nm. It fluoresces orange-red and can be detected using a 562-588 nm band pass filter. Conidial viability was defined by the FSC (forward scatter) and SSC (side scatter) characteristics and the mean fluorescence intensity (MFI) of PI in the treated and untreated conidia. Within scatter parameters, the pulse height vs pulse width plots (FSC vs SSC) are used to identify cells of interest based on size and granularity and to isolate single cells passing through the cytometer, thereby removing any non-single cells (doublets, clumps and debris) (Rowley 2012).
Real-time-PCR assay of ags3 and sph3 expressions
After 48 h static incubation, the fungal cultures were analysed for DNA quantification. Expression of the ags3 and sph3 was quantified by real-time polymerase chain reaction (RT- PCR) assay. RNA was extracted by using Allrep fungal DNA/RNA/protein kit. First strand synthesis was performed from total RNA with Quantitec Reverse Transcription kit (Qiagen, Manchester, UK). Forward and reverse primers (Eurofins Genomics, Germany) related to the genes of interest, ags3 and sph3 and a reference gene, sac7 were used (Table 1). A fragment of the gene encoding ags3 and sph3 was isolated from genomic DNA using PCR with master mix prepared by adding SYBR Green (Merck, Dorset, UK). RT-PCR was performed using a 7500 RT-PCR System (Fisher Scientific Ltd, Loughborough, UK). Fungal gene expression was normalized to A. fumigatus sac7 expression. A comparative threshold cycle (Ct) method was used for relative quantification detecting changes in expression of the genes of interest relative to a reference gene.
Statistical analyses
The SPSS software was used for paired sample T- Test calculation showing data sets that were deemed not significantly different (N.S. > 0.05) and data sets that were significant at different levels: *P ≤ 0.05, **P≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001 (SEM bars are shown for n=3).
Combination treatment of A. fumigatus with triclosan and L-AMB
The MICs for triclosan and L-AMB were calculated as 2 and 1 mg/L, respectively [Supplementary file (S1 and S2)]. In order to investigate triclosan and L-AMB synergistic activity, FICI was adapted as explained in materials and methods and Table 2 shows that a synergistic activity (FICI ≤ 0.5) between the agents could be achieved if triclosan and L-AMB were applied at doses equal or less than 0.6 and 0.2 mg/L, respectively. The minimum inhibition effect, when they were added simultaneously (triclosan dose at 0.6 mg/L plus L-AMB dose at 0.2 mg/L) and sequentially (L-AMB followed by triclosan) revealed 60.5% and 60% viability, respectively (Table 2). This means the minimum inhibition (MIC50; 50% growth inhibition) was not obtained. Sequential addition of triclosan-L-AMB to A. fumigatus cells, first triclosan (0.6 mg/L) followed by L-AMB (0.2 mg/L), showed 48.5% viability (counted as MIC), resulting in FICI = 0.5; hence a synergistic interaction was formed. Therefore, the triclosan and L-AMB MICs were reduced to 30% and 20%, and dropped to 0.6 and 0.2 mg/L, respectively, which present sub-MIC doses in this combination treatment.
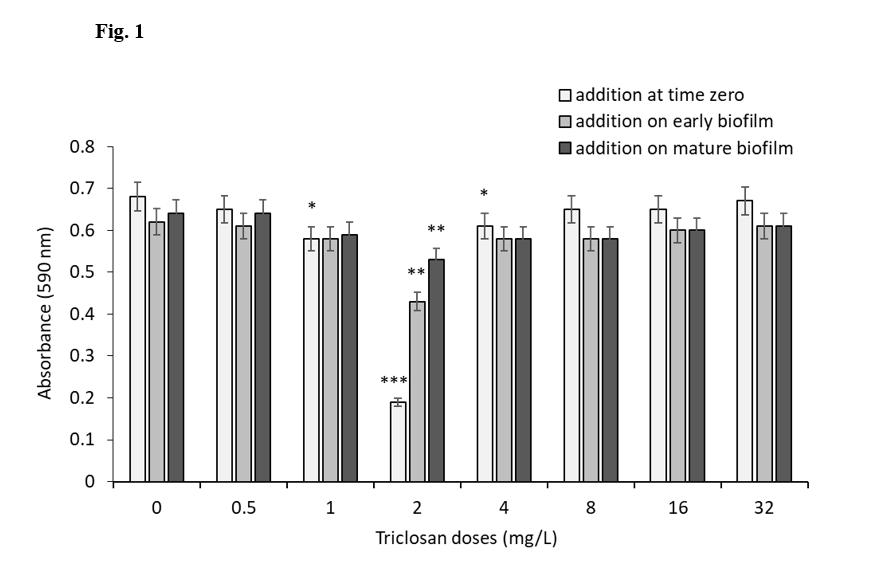
The effectiveness of triclosan and L-AMB alone and in combination against A. fumigatus were analysed (Fig.1).
To confirm checkerboard assay results, various doses of triclosan (0, 0.15, 0.30, 0.60 and 1.20 mg/L) were combined with L-AMB (0.2 mg/L), sequentially (first triclosan followed by L-AMB) and relevant ODs were analysed using Compusyn software (Table 3).
Any CI value between 0 and 1 represent synergism, values equal to 1 shows additivity, and anything above 1 shows antagonism. Among 5 combination data points, 2 represent synergy (CI < 1): triclosan (0.6 and 0.3 mg/L) followed by L-AMB (0.2 mg/L). Together, these data support a synergistic activity between triclosan and L-AMB in A. fumigatus. This synergism could be through triclosan triggering persister cells and make them vulnerable to L-AMB.
Diminishing of metabolic activities has been reported for different microbes treated with triclosan or L-AMB (Kovács et al. 2017; Westfall et al. 2019). Our study showed that in A. fumigatus, a synergistic interaction occurs between triclosan and L-AMB when they were added at their sub-MICs, sequentially (triclosan+L-AMB ).
Structure modifications of biofilm and the viability of conidia in A. fumigatus treated with triclosan and L-AMB
In this study, to assess if the lack of viability in the treated samples compared to the untreated control group was because of the lack of cells attachments to a surface or is because of their death by the agents, we used CLSM, PI-based flow cytometry, and CV-based viability assay. As previously shown in Table 2, single treatment with triclosan and L-AMB at their sub-MICs were not effective against A. fumigatus and the best result happened when triclosan followed by L-AMB at their sub-MICs. So, CLSM was used to compare triclosan and L-AMB (MICs) single treatments with triclosan-L-AMB (sub-MICs) combination treatment (Fig. 2). According to the confocal imaging, the combination of sub-MIC levels of L-AMB and triclosan against A. fumigatus showed a decrease in EPS production and biofilm depth as compared with the L-AMB at its MIC. Triclosan revealed the least biofilm depth and no EPS structure was formed in the samples treated with triclosan MIC. The EPS matrix can encapsulate the released compartments (like eDNA, protein, and carbohydrates), which leak out following cell death.
Our findings showed that the total biomass was higher in L-AMB -treated cultures than in samples treated with triclosan (Fig. 3a), whereas the conidial viability of triclosan-treated conidia (~75% of the control) was less affected than that of L-AMB -treated test groups (~60% of the control) (Fig. 3b, c, and d). CV binds to proteins, polysaccharides and nucleic acids and thus stains the EPS and the microbial cells, so that biofilm viability is not measured by CV (Welch et al. 2012; Kvasničková et al. 2016).
PI-based flow cytometry analysis of the conidial viability (Fig. 4) revealed that the viability was affected the most by triclosan (MFI~52% more than control), and the least by L-AMB (MFI~48% of the control). Persister cells appear to be affected by triclosan but not by L-AMB. So, upon removal of L-AMB (or losing its effectiveness), the persister cells can proceed growing (Fig. 3a).
To sum up, conidial viability was decreased by L-AMB, but viable (attached) conidia were able to form biofilm structures more functional than that in the triclosan-treated samples. As a result, L-AMB had a lower impact on biomass than triclosan (Fig. 3a). In L-AMB -treated cultures, the released eDNA was thus captured in the EPS structure as CLSM results demonstrated (Fig. 2c).
Expression levels of A. fumigatus genes encoding cell wall proteins, α-(1,3)-glucans and GAG
To study triclosan and AMB mechanism of action, expressions of two dominant carbohydrates, galactosaminogalactan (GAG) and α-1,3-Glucan, in A. fumigatus conidia cell wall were studied after treatment with triclosan and L-AMB. RT-PCR results revealed ags3 and sph3 down-regulation in triclosan- and L-AMB -treated conidia (Fig. 5).
Here, we assessed triclosan’s antifungal effect against a filamentous fungus A. fumigatus. The antifungal effect was suggested to be through interrupting the quorum sensing signalling system and hence diminishing EPS/biofilm structure in the fungus.
We applied the combination treatment, as a tool to reduce the effective doses for both triclosan and L-AMB. When triclosan and L-AMB were added at their sub-MICs, sequentially (triclosan+L-AMB), a synergistic interaction occurred in A. fumigatus. The improvement of the antifungal effect of L-AMB on the cells could be due to triclosan’s action on persister cells, which make them vulnerable to L-AMB effect. Persisters can endure drug concentrations much higher than the MIC and represent survivor cells, which are phenotypic variants of the wild-type cells rather than mutants (Al-Dhaheri and Douglas 2009).
In A. fumigatus, L-AMB might not affect the conidial attachment to the substrates, nor the attached cells development proliferation. But, among the cells, most likely non-persister cells or the cells which are not resistant against L-AMB, are killed with L-AMB. The alive cells, though, can grow and form biofilm structure. While in a triclosan-treated sample, no EPS/biofilm structure was formed to capture the released cell components. So, triclosan’s effect might be through its action on the persister cells or through the developmental stage of the cells’ life cycle, where the biofilm structure forms. If the latter concept is the case, a quorum quenching role for triclosan against A. fumigatus could be assumed. Whatever the cause, triclosan is a suitable antifungal agent against A. fumigatus as it does not induce resistance in the fungus.
The conidia surface of A. fumigatus (the rodlet surface) is markedly hydrophobic and is composed of 40% hydrophobic methyl groups (González-Ramírez et al. 2016). In either triclosan- or L-AMB -treated samples, the rodlet structure, if it forms, would be less organized than the rodlet of the untreated strain, which putatively modified the ionic strength of the hydrophobin layer in the triclosan- and L-AMB -treated strains. Absence of the rodlet layer promotes the presence of conidia with more hydrophilicity compared to the conidia of the wild-type strain (Girardin et al. 1999). Therefore, the absence of RodA protein may result in a heterogeneous culture comprising both hydrophobic and hydrophilic conidia. The absence of conidia hydrophobicity may be due to the hydrophilic glycoprotein layer, which is normally secreted as a result of either ags3 or chitin synthase mutants during vegetative growth. Consequently, conidia become hydrophilic upon treatment with triclosan and L-AMB, because of a rodlet layer masked by a hydrophilic layer or modified by the ionic strength of the rodlet layer. It has also been found that α-(1,3)-glucans interact among themselves and are a reason for the aggregation of swollen conidia without the involvement of any protein (Fontaine et al. 2010). Hence the absence of α-(1,3)-glucans affects biofilm formation through interrupting the microcolony formation stage, which initiates with conidial aggregation.
These findings may help increase understanding of the mechanisms underlying triclosan activity against moulds, which, for example, may provide triclosan as a suitable coating supplement for the abiotic surfaces.
Competing Interests and Funding
This work was supported by the University of Westminster Cavendish Scholarship.
Data availability
All data generated or analysed during this study are included in this published article.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Roya Tamimi. The first draft of the manuscript was written by Roya Tamimi and Godfrey Kyazze and Tajalli Keshavarz commented on previous versions of the manuscript and read and approved the final manuscript.
- Al-Dhaheri RS, Douglas LJ (2009) Apoptosis in Candida biofilms exposed to amphotericin B. J Med Microbiol 59:149–157. https://doi.org/ 10.1099/jmm.0.015784-0
- Andrews JM (2001) Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48:5–16. https://doi.org/10.1093/jac/48.suppl_1.5
- Balajee SA, Marr KA (2002) Conidial Viability Assay for Rapid Susceptibility Testing of Aspergillus Species. J Clin Microbiol 40:2741–2745. https://doi.org/ 10.1128/JCM.40.8.2741-2745.2002
- Bamford NC, Snarr BD, Gravelat FN, Little DJ, Lee MJ, Zacharias CA, Chabot JC, Geller AM, Baptista SD, Baker P, Robinson H, Howell PL, Sheppard DC (2015) Sph3 Is a Glycoside Hydrolase Required for the Biosynthesis of Galactosaminogalactan in Aspergillus fumigatus. J Biol Chem 290:27438–27450. https://doi.org/ 10.1074/jbc.M115.679050
- Beauvais A, Maubon D, Park S, Morelle W, Tanguy M, Huerre M, Perlin DS, Latgé JP (2005) Two (1-3) Glucan Synthases with Different Functions in Aspergillus fumigatus. Appl Environ Microbiol 71:1531–1538. https://doi.org/ 10.1128/AEM.71.3.1531-1538.2005
- Beauvais A, Schmidt C, Guadagnini S, Roux P, Perret E, Henry C, Paris S, Mallet A, Prévost MC, Latgé JP (2007) An extracellular matrix glues together the aerial-grown hyphae of Aspergillus fumigatus. Cell Microbiol 9:1588–1600. https://doi.org/ 10.1111/j.1462-5822.2007.00895.x
- Chandrasekar PH, Manavathu EK (2008) Do Aspergillus species produce biofilm? Future Microbiol 3:19–21. https://doi.org/ 10.2217/17460913.3.1.19
- Chou T (2010) Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res 70:440–446. https://doi.org/ 10.1158/0008-5472.CAN-09-1947
- Flemming H-C, Wingender J (2010) The biofilm matrix. Nat Rev Microbiol 8:623–633. https://doi.org/ 10.1038/nrmicro2415
- Fontaine T, Beauvais A, Loussert C, Thevenard B, Fulgsang CC, Ohno N, Clavaud C, Prevost M-C, Latgé J-P (2010) Cell wall α1-3glucans induce the aggregation of germinating conidia of Aspergillus fumigatus. Fungal Genet Biol 47:707–712. https://doi.org/ 10.1016/j.fgb.2010.04.006
- Girardin H, Paris S, Rault J, Bellon-Fontaine MN, Latge´ JP (1999) The role of the rodlet structure on the physicochemical properties of Aspergillus conidia. Lett Appl Microbiol 29:364–369. https://doi.org/ 10.1046/j.1472-765x.1999.00643.x
- González-Ramírez AI, Ramírez-Granillo A, Medina-Canales MG, Rodríguez-Tovar AV, Martínez-Rivera MA (2016) Analysis and description of the stages of Aspergillus fumigatus biofilm formation using scanning electron microscopy. BMC Microbiol 16. https://doi.org/ 10.1186/s12866-016-0859-4. :Article number:243
- Gravelat FN, Beauvais A, Liu H, Lee MJ, Snarr BD, Chen D, Xu W, Kravtsov I, Hoareau CM, Vanier G, Urb M, Campoli P, Al Abdallah Q, Lehoux M, Chabot JC, Ouimet MC, Baptista SD, Fritz JH, Nierman WC, Latgé JP, Mitchell AP, Filler SG, Fontaine T, Sheppard DC (2013) Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal β-glucan from the immune system. PLOS Pathog 9:e1003575. https://doi.org/ 10.1371/journal.ppat.1003575
- Henry C, Latgé JP, Beauvais A (2011) α1,3 Glucans Are Dispensable in Aspergillus fumigatus. Eukaryot Cell 11:26–29. https://doi.org/ 10.1128/EC.05270-11
- Hoang TT, Schweizer HP (1999) Characterization of Pseudomonas aeruginosa enoyl-acyl carrier protein reductase (FabI): a target for the antimicrobial triclosan and its role in acylated homoserine lactone synthesis. J Bacteriol Res 181:5489–5497. https://doi.org/ 10.1128/JB.181.17.5489-5497.1999
- Jones RD, Jampani HB, Newman JL, Lee AS (2000) Triclosan: A review of effectiveness and safety in health care settings. Am J Infect Control 28:184–196. https://doi.org/ 10.1016/S0196-6553(00)90027-0
- Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K (2004) Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett 230:13–18. https://doi.org/ 10.1016/S0378-1097(03)00856-5
- Kovács R, Tóth Z, Nagy F, Daróczi L, Bozó A, Majoros L (2017) Activity of exogenous tyrosol in combination with caspofungin and micafungin against Candida parapsilosis sessile cells. J Appl Microbiol 122:1529–1536. https://doi.org/ 10.1111/jam.13452
- Kvasničková E, Paulíček V, Paldrychová M, Ježdík R, Maťátková O, Masák J (2016) DBM 4057 biofilm formation is inhibited by chitosan, in contrast to baicalein and rhamnolipid. World J Microbiol Biotechnol 32. https://doi.org/ 10.1007/s11274-016-2146-9. :Article number:187
- Lee MJ, Sheppard DC (2016) Recent advances in the understanding of the Aspergillus fumigatus cell wall. J Microbiol 54:232–242. https://doi.org/ 10.1007/s12275-016-6045-4
- Lewis K (2007) Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5:48–56
- Llanos A, François J, Parrou JL (2015) Tracking the best reference genes for RT-qPCR data normalization in filamentous fungi. BMC Genomics 16:71. https://doi.org/ 10.1038/nrmicro1557
- Loussert C, Schmitt C, Prevost M-C, Balloy V, Fadel E, Philippe B, Kauffmann-Lacroix C, Latgé JP, Beauvais A (2010) In vivo biofilm composition of Aspergillus fumigatus. Cell Microbiol 12:405–410. https://doi.org/10.1111/j.1462-5822.2009.01409.x
- Lu L, Li M, Yi G, Liao L, Cheng Q, Zhu J, Zhang B, Wang Y, Chen Y, Zeng M (2021) Screening Strategies for Quorum Sensing Inhibitors in Combating Bacterial Infection. J Pharm Anal: In press https://doi.org/ 10.1016/j.jpha.2021.03.009
- Martínez OF, Rigueiras PO, Pires ÁDS, Porto WF, Silva ON, de la Fuente-Nunez C, Franco OL (2019) Interference with Quorum-Sensing Signal Biosynthesis as a Promising Therapeutic Strategy Against Multidrug-Resistant Pathogens. Front Cell Infect Microbiol 8:444. https://doi.org/ 10.3389/fcimb.2018.00444
- Maubon D, Park S, Tanguy M, Huerre M, Schmitt C, Prévost M, Perlin D, Latgé J, Beauvais A (2006) AGS3, an α(1–3)glucan synthase gene family member of Aspergillus fumigatus, modulates mycelium growth in the lung of experimentally infected mice. Fungal Genet Biol 43:366–375. https://doi.org/ 10.1016/j.fgb.2006.01.006
- Mesquita N, Portugal A, Piñar G, Loureiro J, Coutinho A, Trovão J, Nunes I, Botelho M, Freitas H (2013) Flow cytometry as a tool to assess the effects of gamma radiation on the viability, growth and metabolic activity of fungal spores. INT BIODETERIOR BIODEGRAD 84:250–257. https://doi.org/ 10.1016/j.ibiod.2012.05.008
- Movahed E, Tan GM, Munusamy K, Yeow TC, Tay ST, Wong WF, Looi CY (2016) Triclosan Demonstrates Synergic Effect with Amphotericin B and Fluconazole and Induces Apoptosis-Like Cell Death in Cryptococcus neoformans. Front microbiol 21:360. https://doi.org/ 10.3389/fmicb.2016.00360
- Mowat E, Butcher J, Lang S, Williams C, Ramage G (2007) Development of a simple model for studying the effects of antifungal agents on multicellular communities of Aspergillus fumigatus. J Med Microbiol 56:1205–1212. https://doi.org/10.1099/jmm.0.47247-0
- Mowat E, Lang S, Williams C, Mcculloch E, Jones B, Ramage G (2008) Phasedependent antifungal activity against Aspergillus fumigatus developing multicellular filamentous biofilms. J Antimicrob Chemother 62:1281–1284. https://doi.org/10.1093/jac/dkn402
- Ramage G, Rajendran R, Gutierrez-Correa M, Jones B, Williams C (2011) Aspergillus biofilms: clinical and industrial significance. FEMS Microbiol Lett 324:89–97. https://doi.org/ 10.1111/j.1574-6968.2011.02381.x
- Rowley T (2012) Flow Cytometry - A Survey and the Basics.Materials and Methods2
- Scoffone VC, Trespidi G, Chiarelli LR, Barbieri G, Buroni S (2019) Quorum Sensing as Antivirulence Target in Cystic Fibrosis Pathogens. Int J Mol Sci 20:1838. https://doi.org/ 10.13070/mm.en.2.125
- Seidler MJ, Salvenmoser S, Muller FM (2008) Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. ANTIMICROB AGENTS CH 52:4130–4136. https://doi.org/10.1128/AAC.00234-08
- Tobudic S, Kratzer C, Lassnigg A, Graninger W, Presterl E (2010) In vitro activity of antifungal combinations against Candida albicans biofilms. J Antimicrob Chemother 65:271–4. https://doi.org/ 10.1093/jac/dkp429
- Vanhauteghem D, Demeyere K, Callaert N, Boelaert A, Haesaert G, Audenaert K, Meyer E (2017) Flow Cytometry Is a Powerful Tool for Assessment of the Viability of Fungal Conidia in Metalworking Fluids. Appl Environ Microbiol 83:e00938–17. https://doi.org/ 10.1128/AEM.00938-17
- Weatherly LM, Gosse JA (2017) Triclosan exposure, transformation, and human health effects. J Toxicol Environ Health Part B 20:447–469. https://doi.org/ 10.1080/10937404.2017.1399306
- Welch K, Cai Y, Strømme M (2012) A Method for Quantitative Determination of Biofilm Viability. J Funct Biomater 3:418–431. https://doi.org/ 10.3390/jfb3020418
- Westfall C, Flores-Mireles AL, Robinson JI, Lynch AJL, Hultgren S, Henderson JP, Levin PA (2019) The Widely Used Antimicrobial Triclosan Induces High Levels of Antibiotic Tolerance In Vitro and Reduces Antibiotic Efficacy up to 100-Fold In Vivo. Antimicrob Agents Chemother 63:e02312–18. https://doi.org/ 10.1128/AAC.02312-18
Table 1 Nucleotide sequences of primers used in RT-PCR experiments. They all were provided by Eurofins Genomics, Germany.
|
Gene |
Primer |
Sequence (5' > 3') |
Product Size (bp) |
Reference |
|
sph3* |
Forward |
GGGCATATGTCCAAGGTCTTTGTGCCTCTCTATGTG |
657 |
Bamford et al. 2015 |
|
Reverse |
GGCTCGAGCTATTTTCCCATCAAATCCACAAACTC |
|||
|
ags3** |
Forward |
CGGCAGTCTCTACCTTGGTC |
1700 |
Maubon et al. 2006 |
|
Reverse |
TCGTTCTTCAGCTTGACAGC |
|||
|
Rho GTPase activator (sac7) |
Forward |
AGGAGGATGAAAGTAAAGGACCCC |
159 |
Llanos, François and Parrou 2015 |
|
Reverse |
AAACCCCACACTTGGCGAC |
* Encodes a protein belonging to the spherulin-4 family
**Encodes cell wall α (1–3) glucan biosynthesis
Table 2 Checkerboard assay analysis of A. fumigatus triclosan and L-AMB combination treatment.
|
Triclosan (mg/L) |
0 |
0.15 |
0.3 |
0.6 |
|
L-AMB (mg/L) |
|
|
|
|
|
Simultaneously combination treatment |
||||
|
0 |
100* |
95.5 |
93 |
88 |
|
0.05 |
90.5 |
70 |
66 |
67 |
|
0.1 |
87.7 |
70 |
65.5 |
69 |
|
0.2 |
83 |
81.5 |
73 |
60.5 |
|
Sequential combination treatment (L-AMB following by triclosan) |
||||
|
0 |
100 |
96.5 |
95 |
95 |
|
0.05 |
92.5 |
80 |
77 |
77 |
|
0.1 |
90.7 |
70 |
65.5 |
65 |
|
0.2 |
90 |
85 |
70 |
60 |
|
Sequential combination treatment (triclosan following by L-AMB) |
||||
|
0 |
100 |
90.5 |
89.5 |
80 |
|
0.05 |
95 |
87 |
70 |
67 |
|
0.1 |
93 |
70 |
65 |
60 |
|
0.2 |
93 |
67 |
55 |
48.5 |
* Numbers are presented the cells’ viability (%)
Table 3 CI data for non-constant combination (1:1) of triclosan+L-AMB. CI < 1, = 1, and > 1 indicate synergism, additive effect and antagonism, respectively.
|
Triclosan (mg/L) |
L-AMB (mg/L) |
CI |
|
0.00 |
0.20 |
27.36 |
|
0.15 |
0.20 |
40.50 |
|
0.30 |
0.20 |
0.09 |
|
0.60 |
0.20 |
0.002 |
|
1.20 |
0.20 |
160.27 |
- Fig.1.png
A. fumigatus inoculum was cultured and treated with triclosan doses in RPMI1640 medium at 37°C under static condition. Effective dose of triclosan against A. fumigatus was determined by resazurine- based viability assay. Its MIC was calculated as 2 mg/L, which inhibits the viability by ~ 72% after 24 h of incubation. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001 (SEM bars are shown for n = 3)
- Fig.2.png
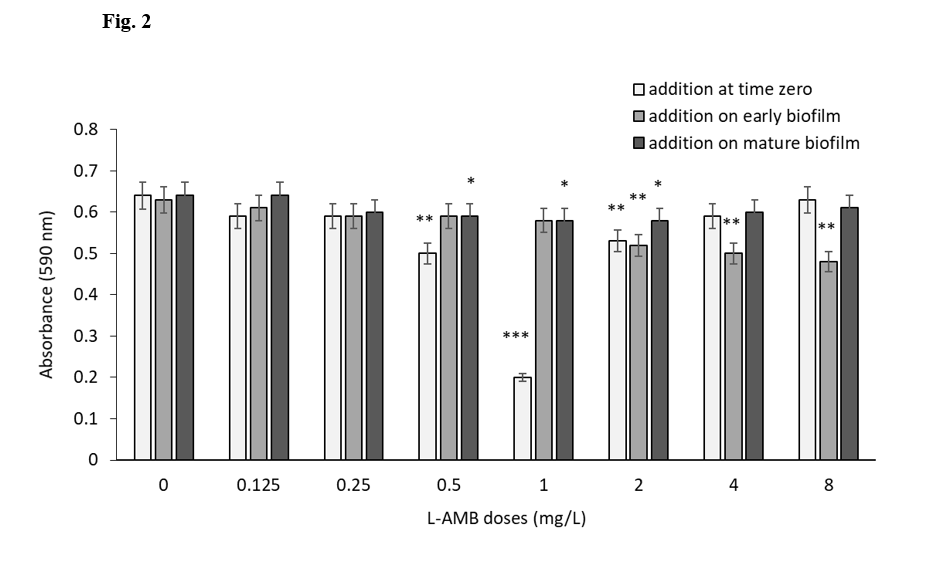
A. fumigatus inoculum was cultured and treated with L-AMB doses in RPMI-1640 medium at 37°C under static condition. Effective dose of L-AMB against A. fumigatus was determined by resazurine- based viability assay. Its MIC was calculated as 1 mg/L, which inhibits the viability by 68% after 24 h of incubation. *P ≤ 0.05 and **P ≤ 0.01 and ***P ≤ 0.001 (SEM bars are shown for n = 3)
{kind=link}
{kind=link}