The phenotypes of individual patients are comprehensively outlined in supplemental Table S1.
Birth History and Family History
Nineteen (67.9%) patients were delivered at full term with no maternal or fetal complications. The mother of P20 was diabetic and had induction of labor at 38 weeks. After delivery, her neonate was admitted to the to the neonatal intensive care unit (NICU) for jaundice and respiratory distress. The mothers of P1 and P4 had pregnancies complicated by pre-eclampsia and immune thrombocytopenia, respectively. P4 was admitted to the NICU for thrombocytopenia. The mother of P3 had gestational hypertension and perception of decreased fetal movements in the 3rd trimester. P3 was admitted to the NICU for hypoxia and concern for streptococcus bacteremia. Two other patients (P13 and P14) were also admitted to the NICU for hypoxia. P5 delivery was complicated by fetal distress, not otherwise specified. The pregnancies of P12 and P16 were complicated by polyhydramnios. P12’s pregnancy was additionally affected by first trimester toxoplasmosis.
P1 and P2 are siblings from one family, and P15 and P16 are siblings from another family. P3, P17, P18, P21, P22, P23, P25, P26, and P27 have siblings or cousins identified as having a neurodegenerative disease or symptoms similar to those manifested in the affected patients but were not available for testing and confirmation. Overall, 15 patients (53.6%) came from consanguineous families.
Developmental History and Disease Presentation
Nineteen patients (67.9%) had at least one delayed developmental milestone prior to the continued developmental regression that characteristically followed. The remaining nine patients reportedly had normal development prior to the regression.
Of the 19 patients reported to have developmental delay, 17 (89.5%) had some form of speech delay; four of these 17 were never able to speak. Sixteen of the 19 patients (84.2%) had some form of gross motor delay. Walking was the most commonly delayed gross motor milestone with 14 patients (87.5%) having some form of delay. Ten out of the 14 patients were never able to walk independently. Of the 16 patients with delayed gross motor milestones, six had a delay in sitting independently (37.5%), four in standing independently (25.0%), four in cruising (25.0%), three in rolling over (18.8%), and one in achieving head control (6.25%).
In addition to speech delay, P21 was reported to have gross and fine motor delay. P13 was reported to have delayed milestones, but no specifics were mentioned in his medical records.
Initial concerns arose when the parents noticed one or more delayed milestones (gross motor delay or a speech delay) or, more commonly, when they noticed developmental regression (with loss of balance being the most frequently cited). The average age at which initial concerns arose was 15 months (median= 14 months, range 6-36 months), with loss of balance being the most commonly reported initial concern (57.7%), followed by developmental delay (34.6%).
Developmental Regression and Disease Progression
Regardless of the presence or absence of developmental delay at an earlier age, all patients eventually experienced some form of developmental regression and lost some or all acquired developmental milestones at varying rates. Gross motor and speech milestones were the first milestones to be affected. Specifically, disturbance in balance (e.g. waddling gait, ataxic gait) was commonly reported by parents to be the first milestone affected (median= 14 months, mean= 16.5 months, range 9-36 months). In patients who never walked, losing the ability to stand (median= 21.5, mean= 25.1 months, range 11-31 months) and the ability to cruise (median= 21 months, mean= 21 months, range 18-24 months) were the most common first milestones to be lost. Overall, patients lost the ability to stand without support at a mean age of 25.1 months (range 11-50 months). Language was the next to be affected at a mean age of 27.6 months (median= 24.5 months, range 12-54 months).
As the disease progressed, patients started losing earlier gross motor milestones, such as crawling (median= 27 months, mean= 30.8 months, range 12-51 months), rolling over (median= 33 months, mean= 35.6 months, range 24-50 months), sitting unsupported (median= 33.5 months, mean= 35.9 months, range 24-57 months), and holding head upright (median= 34 months, mean=53.7 months, range 34-94 months). Fine motor milestones were next to be affected, with a mean age of 47.8 months (median= 30 months, range 14-144 months). Finally, patients experience a gradual deterioration in their bulbar function, starting with choking on thin liquids or drooling (median age= 24 months, mean age= 27.8 months, range 22-39 months), progressing to inability to swallow solid food and being placed on a puréed diet (median age= 41 months, mean= 53.1 months, range= 18-132 months) and eventually requiring placement of a nasogastric tube (median= 6.5 years, mean=6.9 years, range 2.6-12 years) or gastrostomy tube (median=3.8 years, mean=4.8 years, range 2.9-7.5 years). Responsiveness to verbal commands and interaction with parents or examiners were variably affected among patients and were reported to be one of the last milestones to be lost (median= 45.5 months, mean=57.1 months, range 23 months to 15 years).
Six patients (21.4%) died during the course of the study at an average age of 9.9 years (median= 9.9 years, range 6-14.6 years). The causes of death for 2 patients were reported in their charts. P15 was brought to the ER after cardiac arrest at home, recovered normal cardiac rhythm after 14 minutes of CPR, then was intubated and ventilated. DNR (do not resuscitate) status was assigned and she died at the age of 10 years and 9 months (preliminary cause of death: cardiac arrest). P20 was admitted to PICU with respiratory distress (type 2 respiratory failure) that required intubation and ventilation. DNR status was assigned and she died at the age of 14 years and 7 months (preliminary cause of death: cardiopulmonary arrest). The cause of death of the remaining patients was solicited from the parents, the majority of whom reported a “respiratory issue” that preceded the patients’ deterioration and death (Figure 1).
Clinical examinations
Neurological examination revealed axial hypotonia in 12 patients (42.9%) and appendicular spastic hypertonia in 22 patients (78.6%). Eight of the 22 patients with appendicular spastic hypertonia had contractures in one or more joints. Generalized hypotonia and appendicular hypotonia were observed in four (14.3%) and two patients (7.1%), respectively. Seven patients (25.0%) had hyporeflexia on deep tendon reflex (DTR) examination, while 15 patients (53.6%) displayed hyperreflexia. Two patients (P26 and P10) had absent DTR, and one patient (P2) initially had hyperreflexia at 30 months of age followed by hyporeflexia at 35 months. P9 had hyporeflexia limited to the lower limb, while P17 had hyperreflexia limited to the lower limb. Two patients (P18 and P9) had hyperreflexia limited to the upper limb. Four patients (14.3%) were reported to have “ataxia” on physical examination records, but there were 17 patients with loss of balance that may have been the manifestation of undocumented truncal or appendicular ataxia. Similarly, there were also 14 patients with inability to sit independently and 7 with inability to stand independently that may have been manifesting truncal ataxia. Strength assessment revealed lower and upper limb weakness in seven patients (25%), while two patients had weakness limited to their lower limb. P13 and P24 had normal strength in the upper and lower limbs on assessment
Strabismus was observed in eight patients (28.6%), seven of whom also had nystagmus. Overall, nystagmus was evident in 17 patients (60.7%). In addition to nystagmus and strabismus, P5 had amblyopia and anisocoria. P26 had an absent pupillary response. Four patients (14.3%) had intellectual disability, one of whom also displayed aggressive behavior and was diagnosed with ADHD. Other neurological signs that were reported in the physical examination records include positive Babinski sign in three patients, dystonia in three patients, facial dyskinesia in one patient, tongue fasciculations in one patient, dysdiadochokinesia in one patient, and Gower sign in one patient.
Skeletal deformities, including kyphosis, scoliosis, equinovarus, and coxa valga, were observed in ten patients (35.7%). Microcephaly was evident in five patients (17.9%), while relative macrocephaly was observed in 1 patient. Dysmorphology assessment revealed some form of facial dysmorphism, including frontal bossing, prominent forehead, and depressed nasal bridge, in six patients (21.4%).
Review of systems revealed some form of gastrointestinal disease in 12 patients (42.9%), most commonly constipation (75.0% of cases) and gastroesophageal reflux disease (50% of cases).
Disturbed sleep manifesting as nighttime awakening was reported in five patients and as central and obstructive sleep apnea in three patients. Other less common signs and symptoms included emaciation, excessive crying, recurrent infections, reflex autonomic dystrophy, and urinary incontinence.
Of note, serial physical examinations were performed for most patients in the non-Saudi Arabian cohorts. As such, the following calculations apply to the 12 patients from those cohorts. The mean age at which nystagmus was noted is 34.6 months (median= 32 months, range 13-58 months), while strabismus was discovered earlier (mean= 25 months, median= 23 months, range 15-36 months). Increased appendicular tone was revealed on physical examinations at a mean age of 27 months (median= 28.6 months, range =12-58 months), decreased axial tone at 27.2 months (median= 27 months, range= 21-36 months), and muscle weakness at 32.2 months (median= 27 months, range- 23-44 months). DTR examination revealed hyperreflexia at a mean age of 29.7 months (median= 30 months, range= 23-36 months) and hyporeflexia at a mean age of 35.5 months (median= 33 months, range= 25-57 months).
Imaging Studies
Brain magnetic resonance imaging (MRI) was performed on 26 patients (92.9%). MRI was repeated for six of those patients (23.1%). No MRI results were found in the medical records of P2 and P9. The average age at which the first MRI was performed was 3 years (mean= 36.9 months, median= 27 months, range 17 months to 8.6 years).
Variable degrees of cerebellar atrophy were observed in all patients except P4, P16, and P24, who had normal MRI findings at the ages of 1.75 years, 1.42 years, and 3 years, respectively. No repeated MRI were performed for those patients. Two patients had an initially normal cerebellum (P3 at 1.91 years and P17 at 3.83 years), but repeated MRI later showed cerebellar atrophy (P3 at 3 years and P17 at 7.5 years). In our cohort, cerebellar atrophy was noticed on MRI at an average age of 3.36 years (range= 1-8.6 years). In addition to cerebellar atrophy, four patients (P18, P26, P27, and P28) had diffuse cerebral atrophy with widening of cerebral sulci and thinning of corpus callosum noticed at an average age of 4.16 years (range= 3-6 years). There was an increased T2 and FLAIR signal in the periventricular, peritrigonal, and/or parieto-occipital white matter of four patients (16.6%) noted on MRI at an average age of 2.98 years (range= 2.25-4.25 years). Bilateral symmetric altered T2 signal intensities were noted within the globi pallidi of six patients (23.1%), three of whom had hypointense lesions, two had hyperintense lesions, and one was unspecified. Additionally, bilateral symmetric low signal intensity was noted in the substantia nigra and dentate nuclei of P19. Representative MRI images of some of these findings are shown in Figure 2.
Other findings were more variable and included retro-cerebellar fluid collection (arachnoid cyst) in P3, brainstem thinning and atrophy of the pons in P7, nonspecific increased signal intensity at the dorsal aspect of the pons in P22, and atrophic dentate nuclei and symmetrical T2 hyperintense lesions of the head of the caudate nucleus and anterior horn of putamen bilaterally in P23. In addition to cerebellar atrophy, P25 had mild adenoid hypertrophy and mild mucosal sinus disease of ethmoid air cells and maxillary sinuses.
Neurophysiologic Testing
EEG recordings were performed for 18 patients (64.3%). The findings were normal in eight of those patients (44.4%), although four (P8, P14, P16, and P17) had positive seizure history. Three patients (P1, P6, and P27) had slow posterior background rhythm suggestive of bilateral diffuse cerebral dysfunction. Epileptiform discharges were observed in six patients (33.3%), all of whom had positive seizure history. No EEG recording was found for P18, despite having positive seizure history. Overall, 12 patients (42.9%) had at least one reported seizure at an average age of 39 months (range 6 months to 7.3 years). All the reported seizures were generalized, except for two patients (P5 and P16) who had partial seizures. The type of seizure was not reported for two (P15 and P18) of the 12 patients.
Electromyography (EMG) testing was performed on eight patients (28.6%) and revealed motor-more-than-sensory axonal-predominant polyneuropathy or denervation in six of those patients (75.0%). No electrophysiological evidence of axonal or demyelinating polyneuropathy, defect in the neuromuscular junction transmission, or a primary disorder of muscle was found in the remaining two patients (P3 and P20), although the EMG on one of them (P3) showed evidence of L4-L5 radiculopathy.
Nerve conduction velocity (NCV) tests were performed on nine patients (32.1%). These studies revealed distal axonal-type motor neuropathy in seven of those patients (77.8%), while two (P4 and P23) had normal findings.
Eleven patients (39.3%) were tested by flash visual evoked potentials (FVEP), eight (72.7%) of whom showed delayed FVEPs with reduced amplitude bilaterally. Interestingly, electroretinography (ERG) was done on seven of those patients and revealed normal findings in all except P12 who had intense dysfunction of the retina bilaterally compromising the rod system. P10, P11, and P19 had normal findings on FVEP testing.
Brainstem auditory evoked response (BAER) was tested in 16 patients (57.1%). Fourteen patients (87.5%) had an abnormal response signifying some degree of hearing loss. P20 and P23 had normal findings.
Laboratory results
Aspartate aminotransferase (AST) and Alanine Aminotransferase (ALT) levels were quantified in 23 patients (82.1%). All patients had an elevated AST to ALT ratio. P9 had an elevated ALT and normal AST at one point for unknown reasons. Lactate dehydrogenase (LDH) was ordered for 10 patients (35.7%), and it was elevated in all 10.
Other labs that were ordered for some patients include CBC, serum amino acids, lactic acid, ammonia, acylcarnitine profile, urine organic acids, and CK. P28 had high serum cholesterol levels and high arginine levels. CBC revealed slightly elevated platelet count in P27 and low MCV in P25 and P24. The organic acid profile of P25 revealed decanedioic acid and low carnitine. Additionally, this patient and P22 had elevated alkaline phosphatase levels. Plasma amino acid profile of P1 showed mildly elevated glycine. Carnitine was low in P21, who also had elevated serum sodium, chloride, and urea levels. Microcytic hypochromic anemia was reported in P15. Organic acid profile of P7 showed elevated succinic acid and slightly low total carnitine.
A nerve biopsy was taken from P11 around 36 months of age and showed few but very prominent axonal swellings. Intra-axonal inclusions and abnormal mitochondrial morphology were seen in the nerve biopsy of P14. His muscle biopsy showed neurogenic atrophy with intra-sarcoplasmic glycogen. A muscle biopsy from P15 revealed mild to moderate fiber size variation with scattered atrophic change, sub-sarcolemmal glycogen accumulation in multiple fibers, and occasional dark blue fibers on NADH staining and scattered subsarcolemmal mitochondrial accumulation. Neuropathic changes were observed on the muscle biopsy taken from P18.
Molecular Genetic Testing
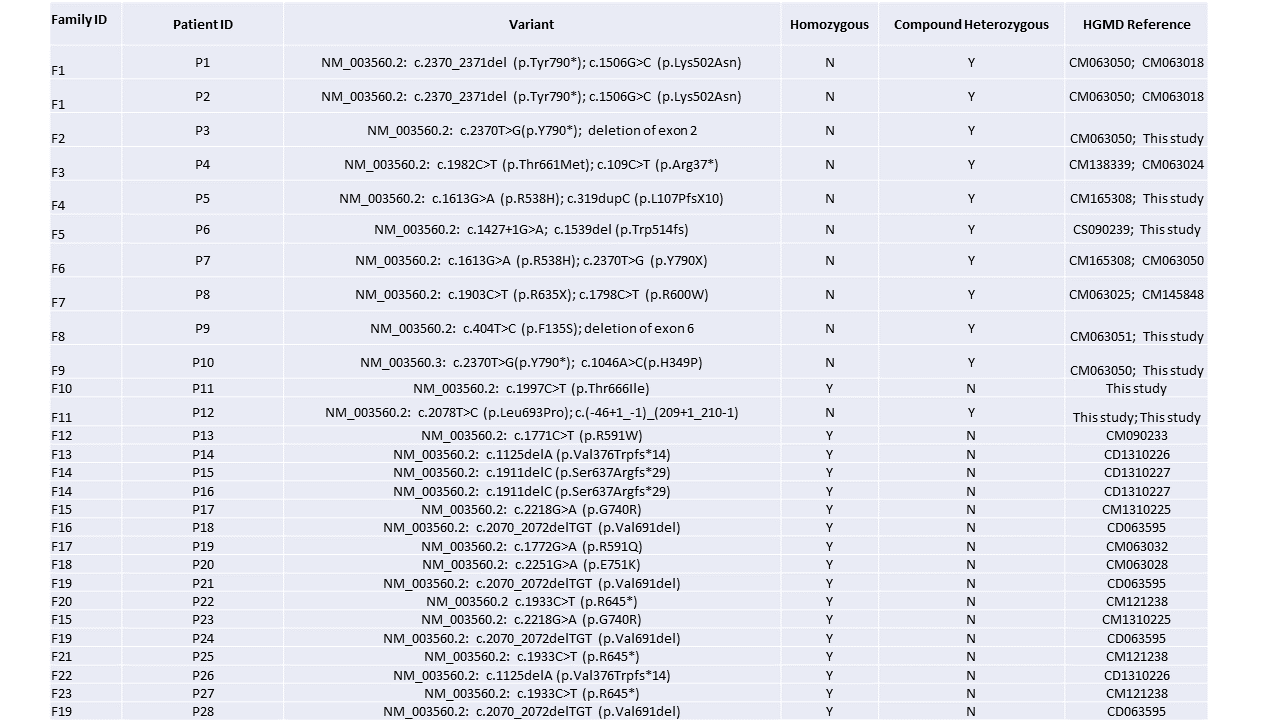
Sequencing of PLA2G6 revealed 11 different homozygous variants and 10 different compound heterozygous pairs for a total of 16 unique individual variants, eight of which were not previously reported in literature. The variants are summarized in Table 1.
All variants for patients in this cohort were initially tabulated and grouped according to the underlying variant type such as truncating or non-truncating (missense and in-frame deletion). Averages for age at initial onset of overall symptoms as well as at initial loss of skills in specific developmental categories were calculated for the entire cohort and for each underlying variant-type. These averages were then graphed to evaluate for potential correlations between underlying variant-type and phenotype.
The variants for all patients in this cohort were subsequently analyzed and common variants were grouped together. Graphs (Supplemental Graphs S1-S5) were then created to reflect the age of onset of initial concern or onset of specific phenotype (i.e. age at loss of skills in specific developmental category) for each variant identified within the cohort. Some patients were plotted twice, if they were compound heterozygous for variants in PLA2G6, to ensure that the potential impact of each variant on phenotype was considered in our analysis. Given that this is a retrospective study, data was not available for every patient in every category. In those cases, nothing was plotted for the respective patients which resulted in a variable number of datapoints plotted on each graph. In addition, if retrospective chart review clearly confirmed that skills within a specific developmental domain were never achieved at all, this was designated as 0 months for age of onset of initial loss of skills in that domain.
Specifically, analysis of our data by underlying type of variant revealed that patients with truncating variants for one or both of their underlying variants had an earlier average age at the time of initial concern and an earlier average onset of initial regression across all developmental domains. This was true when compared to the averages across the entire cohort and when compared to the combined averages for non-truncating variants. This is not entirely unanticipated since truncating variants are typically more deleterious to downstream enzyme function than other variations and thus this was our expected hypothesis. Using a one-tailed T-test we were able to show there was a statistically significant difference when comparing truncating variants compared with non-truncating in the time of initial concern (p = 0.04), initial loss of language (p = 0.001), initial loss of fine motor skills (p = 0.009), and initial loss of bulbar skills (p = 0.007) (Figure 3). We did not observe a statistically significant difference in initial loss of temporal skills (p = 0.11) but note the sample size in both cohorts was smaller (n=8) and thus not adequately powered. The average age for initial concerns to be noted for the entire cohort was 15 months, while for truncating variants it was 13.5 months (n = 16, standard deviation (SD) 3.9 months), for non-truncating variants it was 17.5 months (n = 11, SD 7.7 months). This is consistent with the relatively well-established average age of onset of INAD symptoms between 6 months and 3 years of age.
Mean initial loss of language for truncating variants was 22.1 months (n = 13, SD = 7.2) and non-truncating variants was 36.3 (n = 8, SD = 12.2). Mean initial loss of fine motor skills for truncating variants was 32.8 months (n = 13, SD =17.7) and non-truncating variants was 68.1 months (n = 10, SD = 45.4). Mean initial loss of bulbar skills for truncating variants was 28.7 months (n = 12, SD = 14.6) and non-truncating variants was 61.0 (n = 10, SD = 38.01. Mean initial loss of temporal skills for truncating variants was 40.5 (n = 8, SD = 14.8) and non-truncating variants was 65.6, (n = 8, SD = 53.4).
{kind=link}