3.1 Molecular docking is the most popular CADD method against COVID-19
CADD has seen broad application in drug discovery against COVID-19. In our collected publications, most of them employed docking screen[37–43], molecular simulation[44], pharmacophore models[45, 46], or machine learning-based virtual screens[47] for drug discovery. Docking screen was the most popular one, which took over 96% of the works. They took the advantage of well-established docking tools including the academic software AutoDock[27] or AutoDock Vina[25], DOCK6[48], LeDock[49], rDock[50], LigandFit[51], etc., or the commercial software Glide[52], GOLD[53], Surflex-Dock[54], MOE[55], Discovery Studio[56], etc. A typical docking screening conducted by those studies includes three steps: 1) molecular docking, to predict binding modes and affinities of each molecule in a compound library; 2) analyzing ADMET to predict the pharmacokinetic properties; 3) molecular dynamics, to exam the binding stability and free energy. Besides the docking methods, compound libraries were also a key factor for virtual screening. They mainly included FDA-approved, investigational and experimental drug libraries, natural product libraries, and comprehensive compound libraries, e.g. ZINC[23], ChEMBL[57], PubChem[22]. Particularly, almost one-third of our collections employed the FDA-approved, investigational, or experimental drug libraries, indicating the importance of drug repurposing in dealing with this pandemic.
A successful CADD study not only depends on computational methods but also relies on the target proteins. In our collected publications, we observed 16 drug targets used for drug design (Table 1 and Table S1). They included the 12 proteins from SARS-CoV-2 but also 4 proteins from human, a host of the virus. The former included the viral non-structural proteins, such as nsp1~16 involved in the viral pathogenicity, replication, modification of viral RNA, or assembly of virions, and structural proteins, such as spike protein (S) related to the viral infection, or nucleoprotein (N) related to viral assembly. The latter included 4 enzymes including angiotensin-converting enzyme 2 (ACE2), transmembrane protease serine 2 (TMPRSS2), dihydroorotate dehydrogenase (DHODH) and proteinase-activated receptor 1(PARP-1). In all those 16 proteins, 15 had at least one experimentally solved structure, except nsp14. Particularly, as of August 4, 2021, the number of experimental structures for nsp3 (PLpro), nsp5 (Mpro), and S are as many as 308, 369, and 466, respectively in the PDB[28], which are remarkably more than those for the other targets (Table 1 and Table S1). Comparative analysis showed that some of the proteins showed significant conformational changes upon ligand binding. For example, the PLpro-inhibitor complex and PLpro apo structure were remarkably different at BL2 loop (Figure S1a). And open and closed conformations were observed in the exterior residues (e.g. Q24, D30, E35, E37, D38, Y41, Q42, N53, E56, Y83, Q325, E329, Q330, K353, R393) of ACE2 (Figure S1b). Those structural variations have been carefully investigated in some of the CADD studies[58, 59].
Table 1
The main drug targets for the treatment of SARS-CoV-2 used in CADD studies.
| Targets | Short name | Organism | Solved structures (Solved complexes) |
| Host translation inhibitor nsp1 | nsp1 | SARS-CoV-2 | 21 (0) |
| Non-structural protein 3 | nsp3, PLpro | SARS-CoV-2 | 308 (262) |
| 3C-like proteinase | nsp5, 3CLpro, Mpro | SARS-CoV-2 | 369 (177) |
| Non-structural protein 9 | nsp9 | SARS-CoV-2 | 12 (0) |
| RNA-directed RNA polymerase | nsp12, RdRp | SARS-CoV-2 | 28 (9) |
| Helicase | nsp13, Hel | SARS-CoV-2 | 67 (58) |
| Proofreading exoribonuclease | nsp14, ExoN | SARS-CoV-2 | 11 (0) |
| Uridylate-specific endoribonuclease | nsp15 | SARS-CoV-2 | 44 (26) |
| 2'-O-methyltransferase | nsp16 | SARS-CoV-2 | 28 (24) |
| Spike glycoprotein | S | SARS-CoV-2 | 466 (13) |
| Nucleoprotein | N | SARS-CoV-2 | 21 (0) |
| Angiotensin-converting enzyme 2 | ACE2 | Homo sapiens | 58 (1) |
| Transmembrane protease serine 2 | TMPRSS2 | Homo sapiens | 1 (1) |
| Dihydroorotate dehydrogenase | DHODH | Homo sapiens | 79 (75) |
| Proteinase-activated receptor 1 | PAR-1 | Homo sapiens | 5 (1) |
| Note: Solved structures refer to the number of solved 3D structures in the PDB; Solved complexes refer to the number of solved 3D structures complexed with ligands (binding in the active sites) in the PDB. |
3.2 CADD studies predicted diverse compounds against COVID-19
Since the outbreak of SARS-CoV-2, CADD has been widely used either by drug repurposing or screening novel compounds. In the 181 research reports collected, we obtained a total of 1073 compound records, which included molecular sources, research methods, targets, active sites for binding, etc. Among these compounds, 53.6%, 11.4%, 7.5%, 7.1%, 6.6% target Mpro, S, RdRp, PLpro and ACE2 respectively (Figure 1a). Some of the compounds have been recommended by multiple research teams. For example, lopinavir, one of the compounds recommended by nine virtual screening researches, has been proved to inhibit SARS-CoV-2 with the IC50 of 9.12 µM in the later wet-lab research[60]. Thus, we summarized the compounds recommended more than twice in targeting a receptor, as shown in Figure 1b, suggesting their potential in the following research.
After eliminating the redundant records, we finally obtained 779 recommended compounds, including 457 (59%), 114 (14.6%), 74 (9.5%), 70 (9%) and 69 (8.9%) compounds targeting Mpro, S, PLpro, RdRp, ACE2 and the others respectively (Table 2). We also observed that a target protein may contain two or more binding pockets for drug design. These targets include nsp1, PLpro, nsp9, nsp13, nsp14, nsp16, S protein, N protein and ACE2 (Table S2). The different pockets usually play different roles in a target protein. For example, in the papain-like protease domain of PLpro, the S1/S2 pocket is the binding regions of catalytic sites while S3/S4 pocket (the BL2 loop) is substrate-binding related sites[58]. And in nsp13, both the ATP binding pocket and RNA binding pocket are used for screening[61]. In this work, we did not distinguish the detailed impact of the binding pockets, but performed the analysis by comparing the recommended compounds using the same binding pockets but not the same target proteins.
In order to compare compounds obtained from different studies, we performed unified molecular docking against the top five popular drug targets, including 457 recommended compounds against the catalytic site of Mpro, 73 recommended compounds against the PPI surface of S, 68 recommended compounds against the catalytic site of RdRp, 68 recommended compounds against S3/S4 pocket of PLpro and 41 recommended compounds against the catalytic site of ACE2 (Figure 2a). The unified docking was compared to the nine experimentally validated compounds against Mpro, with the corresponding docking scores of ~ -7.0 kcal/mol (Table S3). We observed that 76%, 75%, 47%, and 59% recommended compounds in Mpro, S3/S4 pocket of PLpro, RdRp, and S respectively achieved lower docking scores (lower than -7.0 kcal/mol), which suggest stronger binding. Particularly, GRL0617, the ranking-first compounds targeting PLpro S3/S4 pocket (docking score = -10.0 kcal/mol), was confirmed to have strong affinity (KD = 1.93 µM)[58], after predicting in July 2020[37]. Unlike the four targets, only 2% recommended compounds against ACE2 achieved scores were lower than -7.0 kcal/mol. It could be attributed to the flat interface between ACE2 and compounds, which originally binds with the viral S protein. Hence it is much more challenging to develop drugs against ACE2 than the others.
We also evaluated the drug-likeness of the recommended compounds using Lipinski’s “Rule of Five”[62]. The results showed that 67% and 81% of compounds targeting Mpro, PLpro respectively met at least four of the Lipinski‘s rules (Figure 2b). However, only 53%, 56%, 57% of compounds targeting S, ACE2, and RdRp respectively were able to fulfill the criteria, implying the difficulties to discover qualified drugs against those targets.
Table 2
Statistics of studies on the SARS-CoV-2 inhibitor discovery using CADD.
| Target | #Refs | #Hits | Target | #Refs | Hits |
| nsp1 | 2 | 14 | nsp16 | 2 | 25 |
| PLpro | 12 | 74 | S | 34 | 114 |
| Mpro | 124 | 457 | N | 5 | 18 |
| nsp9 | 2 | 9 | ACE2 | 16 | 69 |
| RdRp | 23 | 70 | TMPRSS2 | 6 | 29 |
| Helicase | 2 | 5 | DHODH | 1 | 27 |
| ExoN | 2 | 2 | PAR-1 | 1 | 2 |
| NendoU | 2 | 12 | | | |
3.3 Comparison studies between CADD compounds and co-crystallized inhibitors
So far, a large number of co-crystal structures of SARS-CoV-2 targets in complex with inhibitors are available in PDB, for example, 136, 74, 37 and 16 compounds are co-crystallized with Mpro, DHODH, ATP binding domain of nsp13, and papain-like protease domain of PLpro respectively (Tables S4). It is a valuable source for evaluating and improving the CADD recommended compounds by comparing with those experimentally determined inhibitors. In the following sections, we will elaborate the results of our comparison studies on molecular similarity, drug-likeness, and binding modes for those compounds.
3.3.1 Molecular similarity
We first analyzed molecular similarity between the compounds discovered by CADD and the co-crystallized inhibitors in terms of 2D (morgan fingerprint) and 3D (LS-align) similarities, which were evaluated by Tanimoto coefficients and PC-score respectively. The result showed that in the available inhibitors against 11 SARS-CoV-2 targets, the average Tanimoto coefficients were all less than 0.3, indicating that the majority of CADD compounds were dissimilar to the co-crystallized inhibitors (Figure 3a). In other words, CADD compounds showed diverse chemical scaffolds compared with the co-crystallized inhibitors. Nevertheless, we also found that 19, a small number of CADD compounds were highly similar to the Mpro inhibitors (Tanimoto similarity coefficient ≥ 0.5) (Table S5). Among them, 14 flavonoid analogues shared the same skeleton as the known inhibitors Myricetin[63], 7-O-methyl-myricetin[63], 7-O-methyl-dihydromyricetin[63] and Baicalein[64]. In addition, Perampanel is the lead compound of the known inhibitor COMPOUND4[65], which also has the same skeleton structure. Flovagatran, DB04692, Compound 23727975 and Caspase-1 Inhibitor VI are peptoid compounds, which have the most similar topological structures with the known inhibitor MPI1[66], MPI6[66], N3[67] and Z-VAD(OMe)-FMK[68] respectively. Compared to the Tanimoto coefficients, the 3D similarity analysis showed higher similarities that the average PC-scores are all larger than 0.5. And they were even over 0.7 for the compounds targeting Mpro, nsp13 and DHODH, which indicated that the compounds shared rather similar topology in space despite of the different chemical composition (Figure 3b). What is more, 1.5%, 42.4%, 5.9%, 80%, 2.4%, 6.9% and 11.1% CADD compounds targeting PLpro (S3/S4 pocket), Mpro, RdRp, nsp13 (ATP binding site), ACE2 (catalytic pocket), TMPRSS2 and DHODH respectively, showed that maximal PC-score were greater than 0.8. The above results illustrated that the CADD methods not only potentially discovered the variants of co-crystallized inhibitors but also created diversities for further drug discovery.
3.3.2 Drug-likeness
We further performed the comparative study on the drug-likeness of the CADD compounds with the known co-crystallized inhibitors against the corresponding targets in terms of cLogP (calculated lipophilicity), HBA (hydrogen-bond acceptor), HBD (hydrogen-bond donor), MW (molecular weight). In this section, we investigated the compounds targeting Mpro (136 inhibitors, 457 CADD compounds), PLpro S3/S4 pocket (13 inhibitors, 68 CADD compounds) and DHODH (74 inhibitors, 27 CADD compounds) because they have multiple co-crystallized structures, making the comparative analysis more compelling. According to Lipinski’s “Rule of Five”, the compounds with MW ≤ 500, HBA ≤ 10, HBD ≤ 5 and -2 ≤ cLogP ≤ 5 are predictive of good permeability and absorption[62]. Because the latest evaluation of 204 small molecule oral drugs by Brown et al.[69] showed 30% approved drugs with an MW higher than 500 Da. Therefore, we raised the MW standard in "Rule of Five" to 600 Da in the following analysis. The chemical space comparison between SARS-Cov-2 Mpro inhibitors and CADD compounds was illustrated in a tSNE map (Figure 4). 54% of the CADD compounds met the "Rule of Five". Moreover, the distribution of drug-likeness features was basically the same as that of Mpro inhibitors. The clusters of inhibitors showed that the peptoid inhibitors represented by PF-00835231[70], MPI7[66], and Narlaprevir[71] accounted for the majority (52%) of the currently known Mpro inhibitors. These inhibitors typically had greater MW and binding affinities, which was closely related to the larger binding pocket of the active site in Mpro[65, 70, 72]. More significantly, a cluster of CADD compounds presented in the region at which peptoid inhibitors was distributed, implying their similar drug-likeness properties. The remaining 46% of CADD compounds violated one or more rules in the "Rule of Five". Some of them showed higher hydrophilicity, and the others had larger molecular weights (MW>600) or higher lipophilicity (cLogP>5). For PLpro and DHODH (Figure S2), 78% of CADD compounds targeting PLpro S3/S4 pocket met the "Rule of Five" and had a more diverse chemical space than the known inhibitors. All the CADD compounds of DHODH met the "Rule of Five" and fell within the chemical space of known inhibitors.
3.3.3 Binding modes
To reveal the molecular mechanism, we next compared the binding modes of the CADD compounds with the co-crystallized inhibitors. Our analysis was focused on the 19 CADD recommended compounds of Mpro described above, which shared highly similar topological structures with co-crystallized inhibitors. We compared the docking conformation of these CADD compounds to the corresponding co-crystallized structures (Table S5) in terms of the chemical compositions for favorable binding. Among the 19 recommended compounds, Perampanel, an anti-epileptic drug, possesses a distinctive topology. And compared with the co-crystallized inhibitor ( named COMPOUND4 in Zhang’s report[65]), Perampanel lacked the polar interaction with His163 and Glu166 and hydrophobic interaction with the S2 pocket in its predicted binding mode (Figure 5a). Moreover, the binding assays also showed that the activity of Perampanel was much lower than that of COMPOUND4 (e.g., IC50 of 100~250 µM vs. 4.02 µM[65]). 14 flavonoid analogues shared the similar binding mode and key interactions with the co-crystallized inhibitors. For example, the parent nucleus of Baicalin docked into the Mpro pocket in a similar manner to a co-crystallized inhibitor Baicalein, which possessed approximately 7-fold higher affinity than Baicalin (e.g., IC50 of 0.94 µM vs. 6.41 µM[64]). Since the 7-O-glucuronide was unable to fit in the S1' pocket appropriately in this binding mode, thus it resulted in the weaker polar interactions with Leu141, Gly143 and Glu166. However, the interaction of 7-O-glucuronide with Thr24-26 on the outside of the S1' pocket contributed to the binding of Baicalin (Figure 5b). As another example, Quercetin and Robinetin shared the similar binding mode with the covalent inhibitor Myricetin (IC50 of 0.63 µM[63]), and both could form polar interactions with GLU166 (Figure 5c and Figure 5d). Additionally, since the pyrogallol group in Myricetin worked as an electrophile to covalently modify the catalytic cysteine of Mpro[63], it can be speculated that the pyrocatechol group in Quercetin and the pyrogallol group in Robinetin may also have the ability to covalently bind to catalytic cysteine. In-depth study of these flavonoids will further promote the design of non-peptidomimetic covalent or non-covalent inhibitors against Mpro. The remaining four compounds are peptoids. Among them, DB04692 and Compound 23727975 shared the similar non-covalent docking modes, with identical or similar groups binding in the S1 and S2 pocket as that of co-crystallized inhibitors (Figure 5e and Figure 5f). Moreover, both had a reactive warhead in the S1' pocket, close to Cys145, implying the ability to form covalent bond.
3.4 An atlas of the inhibitors against SARS-CoV-2
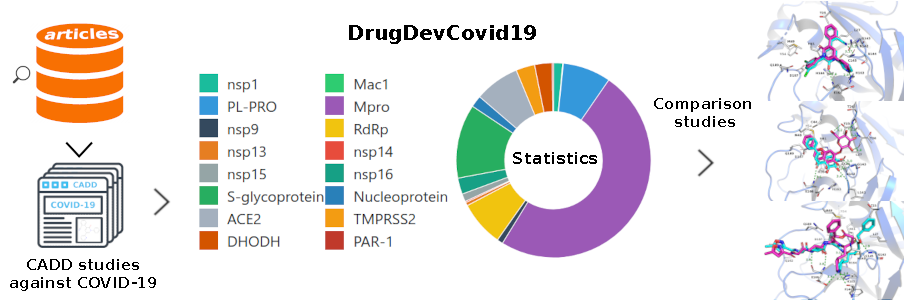
To facilitate the analysis of the discovered drug candidates against SARS-CoV-2, we constructed an online database, named DrugDevCovid19, which collected the compound structures, target protein structures, chemical properties, screening methods, experimental data, and 3D docking modes from the primary published computational and experimental reports. The current version contains 779 compounds and 16 drug targets extracted from 181 reports mentioned above. It is publicly available via a user-friendly interface at http://clab.labshare.cn/covid/php/index.php (Figure 6). Compounds and targets of interest can be searched in a variety of ways at the above webpage. The ‘Entries List’ presents all the compound records retrieved from literature. The records were classified by the drug target and displayed in a donut chart. Users can click the regions in the chart to view the detailed records. The ‘Candidates List’ presents all the non-redundant compounds contained in the records. Similarly, users can click each region to view the compounds which are sorted by record number. Since there may be multiple druggable sites on one target, the compounds were further classified according to the active site and sorted by the binding affinity values predicted by AutoDock Vina[27]. The ‘Targets List’ presents all the potential drug targets. For each target, the web interface illustrates the full name, PDB code(s), the potential active sites retrieved from literature, the organisms, and the Uniprot ID. Users can click the target name to view more information, such as the target function retrieved from UniProt and the surface residues in the active site. The 3D structural features of the active site can be viewed via an NGL Viewer. For each CADD compound, we showed its chemical spatial features, drug function (retrieved from DrugBank if it exists), structural information (including SMILES, 2D, and 3D structures), and Vina scores, with an interactive 3D visualization of the docking conformation.
{kind=link}