PDGFRα + S-MSCs readily differentiated into brown-type adipocytes in vitro
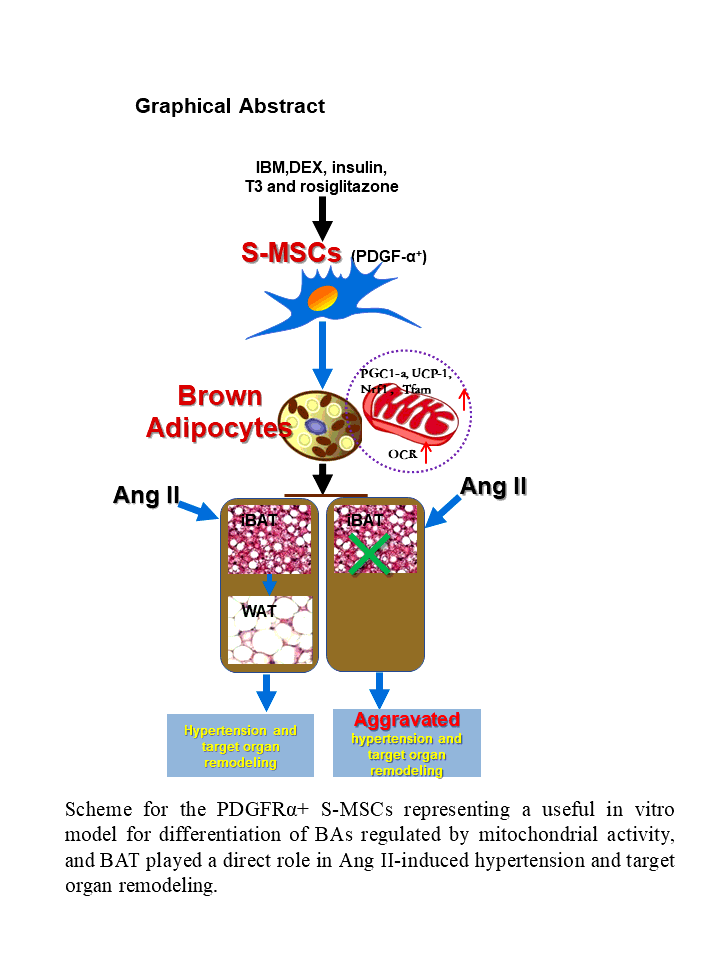
BAs are known to be derived from the multipotent progenitor cells, such as PDGFRα+ ASCs [10]. S-MSCs are a type of easily attainable MSCs recently favored in stem cell research and the development of tissue therapies [12–15]. We reasoned that, S-MSCs could also be induced to differentiate into BAs. Here, we demonstrated that S-MSCs isolated from the dermis of mouse skin expressed the stem cell markers CD90/105 and PDGFRα by FACS analysis (Fig. 1A). In order to initiate BA induction, S-MSCs were stimulated with an adipogenic cocktail containing insulin, IBMX, dexamethasone, triiodothyronine (T3), and rosiglitazone for the indicated periods. Lipid droplets became discernible in the cells with 2 days of stimulation and could be observed in about 90% of the cells by day 8, indicating the transition of the cells into BAs (Fig. 1B). Thermogenic markers mitochondrial UCP-1 and PGC-1α have been dubbed the hallmark of BAs [9]. We measured the expression of UCP-1 and PGC-1α in the cells during the course of induced differentiation by RT-PCR and found that the levels of UCP-1 and PGC-1α transcripts steadily increased during the 10-day period of differentiation, with a slight drop in PGC-1α level at day10 (Fig. 1C). This mRNA expression pattern was largely correlated with their protein levels during differentiation, in which both markers showed a continual increase with a minor drop on day 10 (Fig. 1D). These results showed that S-MSCs expressed PDGFRα and readily differentiated into BAs upon adipogenic induction in vitro.
BA differentiation of S-MSCs is accompanied by enhanced oxygen consumption
Increased mitochondrial activity is a prerequisite for MSC differentiation into adipocytes [17]. Using the Seahorse XFe96 analyzer, we found that oxygen consumption of S-MSCs was highly increased during the differentiation periods from day 2 to day 8 (Fig. 2A), indicating that BA differentiation of S-MSCs is a process that requires enhanced mitochondrial function and energy supply. Mitochondrial function as indicated by ATP levels and mitochondrial complex activities such as basal mitochondrial respiration, proton leak, maximal respiratory capacity, and spare capacity were also significantly enhanced during the adipogenic differentiation process (Fig. 2B-F), suggesting that S-MSCs not only acquire higher baseline oxygen consumption, but also exhibit more activated mitochondrial function during BA differentiation.
Mitochondrial biogenesis was increased during BA differentiation of S-MSCs
To further examine the role of mitochondria in brown adipogenic differentiation of S-MSCs, we measured mitochondrial mass in the S-MSCs stimulated with brown adipogenic cocktail for the indicated times by staining with MTGFM and subsequent analysis by flow cytometry. Results demonstrated that mitochondrial mass was significantly increased in the S-MSCs after 2 days of BA differentiation and remained at a relatively high level throughout the BA differentiation process (Fig. 3A). Consistently, fluorescence microscopy of cells stained with MTGFM and DAPI also showed the increased mitochondrial content in cells at day 4 after stimulation compared with unstimulated S-MSCs (Fig. 3B). Immunoblot analysis showed that expression of mitochondrial complex-1 and complex-4 proteins was robustly enhanced during the BA differentiation process (Fig. 3C). Further, real-time RT-PCR revealed that the mRNA levels of mitochondrial TFAM and NRF1, the key regulating factors of mitochondrial biogenesis, were up-regulated significantly during the BA differentiation of S-MSCs (Fig. 3D). These data confirmed that the mitochondrial biogenesis is boosted during the BA differentiation of S-MSCs.
Change of interscapular brown fat tissue to white fat tissue in Ang II-induced hypertensive mice
BAT is mainly composed of BAs. Animal studies has displayed that transgenic ablation of BAT is associated with systemic hypertension [3]. However, the change of BAT in Ang II-induced hypertensive mice is rarely investigated. Here, we employed the Ang II-induced hypertensive mouse model. During 28 days of Ang II infusion at 750 ng/kg/min administered by ALZET osmotic pumps, the Ang II-infused mice showed a significant increase of systolic blood pressure (SBP) as well as remarkable vascular injury compared with the control mice (Fig. 4A and 4B). Subsequent histological analysis revealed extensive brown-to-white adipose transformation in the iBAT extracted from the Ang II-infused mice compared with the control mice (Fig. 4C). For further in vitro assessment, we used Ang II to stimulate BAs either induced from S-MSC differentiation or isolated from the iBAT of mice, and found that expression of brown fat-specific marker UCP-1 and adipogenic marker PGC-1α was decreased in a dose-dependent manner after Ang II stimulation (Fig. 4D and 4E). Together, these data suggest that Ang II causes the whitening of BAT in hypertensive mice and induced BA dysfunction in vitro.
Brown adipose tissue-deficiency enhanced Ang II-induced hypertension and vascular remodeling
Prompted by these results, we next aimed to determine the direct role of BAT in blood pressure (BP) regulation by using iBAT-deficient mice (generated by surgically removing the iBAT depot in C57BL/6 mice and allowing to recover for 6 days, -BAT) and WT mice. At day 7, -BAT and WT mice were further administered Ang II or 0.9% NaCl infusion at 750 ng/kg per minute for 28 days and BP was monitored using the noninvasive tail cuff method. We found that iBAT-deficient mice manifested drastically increased SBP elevation compared with WT mice (Fig. 5A). The histological analysis of aorta sections was used to examine the aorta structural changes, and HE staining showed that 4 weeks of Ang II infusion caused hypertrophy of aortas (intima and media), which was aggravated in iBAT-deficient mice (Fig. 5B). The aortic intima and media thickness were quantified (Fig. 5C). Ang II-induced fibrosis of heart was significantly aggravated in iBAT-deficient mice compared with WT mice (Fig. 5D). Collectively, these data suggested that the direct deficiency of iBAT was able to facilitate Ang II-induced BP elevation and target organ damage.
{kind=link}