MicroRNA target analysis
Three microRNA target prediction databases including TargetScan (release 7.2), miRDB (release 6.0) and microT-CDS (release 5.0) were used to identify potential miR-141-3p and miR-200a-3p target sites in genomic sequences [46–48]. Moreover, 115 genes strongly correlated with Th17 cell differentiation identified through literature mining in our earlier study [49]. The common predicted targets for both microRNAs from Th17-related gene list, TargetScan with a total context + + score ≤ − 0.3, miRDB with a score ≥ 90 and microT-CDS with a miTG score ≥ 0.9 were retrieved using VENNY 2.1 tool [50]. These strict thresholds were chosen to achieve high-confidence target genes and reduce the incidence of false positives. Experimentally validation of predicted microRNA-mRNA interactions was also checked from miRTarBase (release 7.0) and TarBase (release 8.0) [51, 52]
Molecular Signaling Pathway Enrichment Analysis
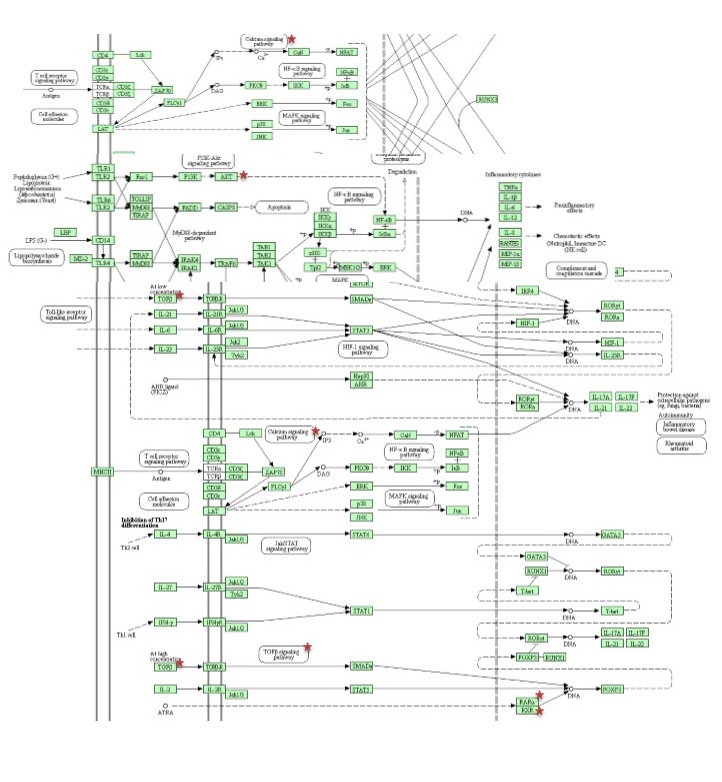
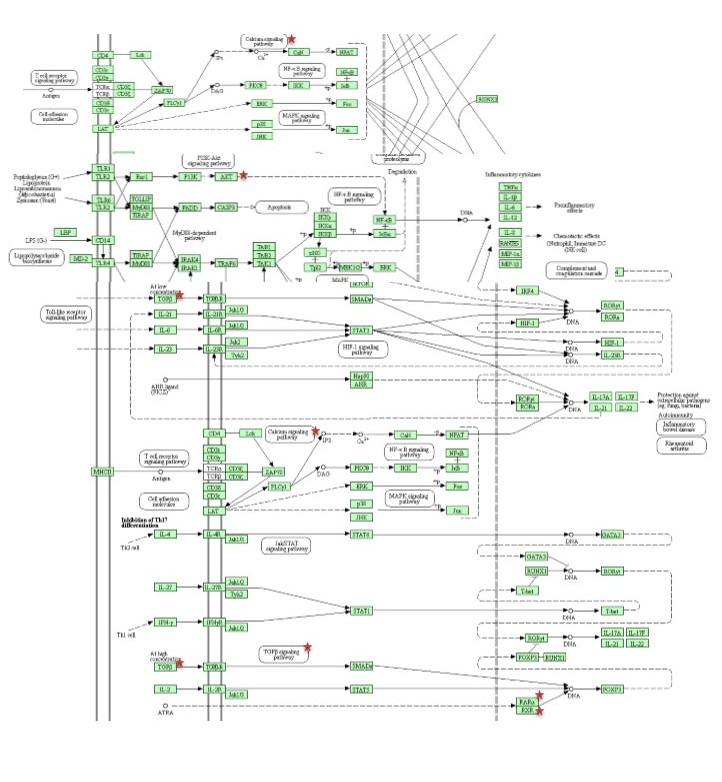
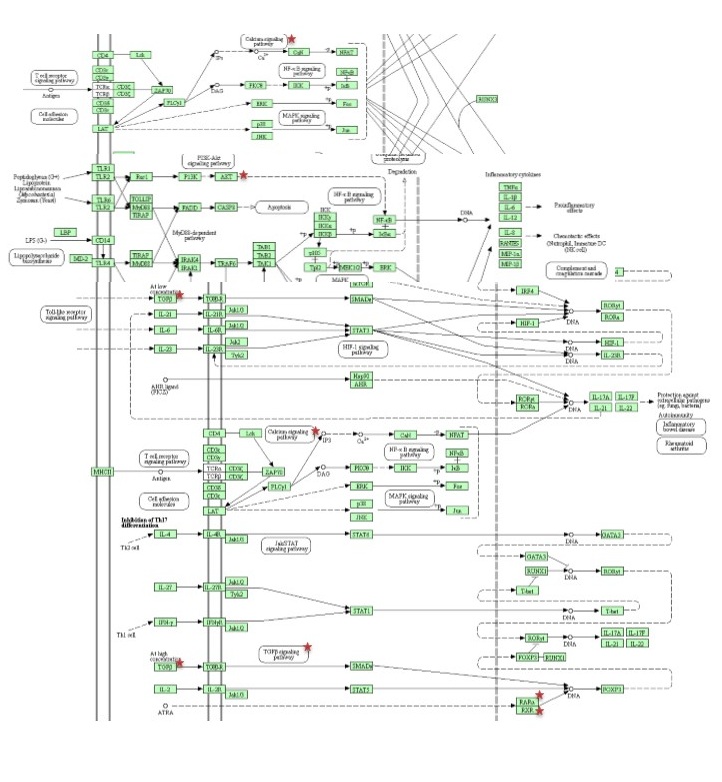
Reactome, KEGG and BioPlanet were employed to obtain a comprehensive view of statistically significant signaling pathways and molecular networks related via predicted target gene (p value < 0.05) [53–55].
Interaction Network Construction
For further evaluating our candidate target gene, we also constructed a gene-based network which included two sets of 55 genes highly associated with autoimmune diseases (gda score > 0.2) and 115 genes strongly involved in Th17 cell differentiation obtained from DisGeNET database (release 7.0) and our earlier study respectively [49, 56]. Interactions of candidate target gene with the genes of both groups were assessed by STRING-db and visualized by Cytoscape software (release 3.8.0) [57, 58].
CD4+ naïve T cell purification
Human peripheral blood mononuclear T cells (hPBMCs) were separated from 20 mL freshly collected, heparinized peripheral blood via Ficoll– Hypaque density gradient centrifugation (Sigma-Aldrich, USA). Purification of naïve CD4+ T cells was performed using magnetic cell sorting system (MACS) according to Naïve CD4+ T Cell Isolation Kit II (Miltenyi Biotec, Germany) instruction. Briefly, non-naïve T cells were magnetically labeled with a cocktail of biotin-conjugated antibodies and anti-biotin microbeads. Isolation of pure naïve CD4+ T cells was achieved by depletion of magnetically labeled non-target cells.
Flow Cytometry
Purification analysis of isolated naïve CD4+ T cells was carried out using flow cytometry. After single cell preparation, FITC-conjugated antiCD4 and PE-conjugated anti-CD45RA antibodies (all antibodies were purchased from eBioscience, USA) were added to cell suspension and resulting mixture incubated for 30 min at 4 °C. Flow cytometry analysis was performed by Canto II (BD FACS Calibur) and analyzed using BD CellQuest Pro software.
Th17 Cell Differentiation
For Th17 cell differentiation, the CellXVivo Human Th17 Cell Differentiation Kit (R&D Systems, USA) was used. Purified naïve CD4+ T cells were cultured at a density of 2 × 105 cell per well in 12-well plates with beads coated with anti-CD3 and containing X-VIVO™ 15, Serum-free hematopoietic cell medium (Lonza, Switzerland), under Th17 cell polarizing condition prepared according to the kit manufacturer’s instruction. Cells were maintained in a humidified incubator containing 5% CO2 at 37 °C for six days and differentiation media were refreshed every other day.
Enzyme linked immunosorbent assay (ELISA)
Th17 cells differentiation was confirmed using Quantikine ELISA Human IL-17 Immunoassay kit (R&D, USA) according to the manufacturer’s protocols. IL-17A content of supernatant culture media was measured 5 hours, 2 days, 4 days, and 6 days after induction of Th17 differentiation. Results were read by microplate reader (ELX 800) at 450 nm in duplicate.
RNA extraction, cDNA synthesis and real-time quantitative PCR (RT-qPCR)
Cells were lysed by TRIzol (Invitrogen, USA) for RNA isolation on day 0, 2, 4 and 6 of Th17 differentiation process. The RNA quality and quantity was determined via a NanoDrop 2000/2000c spectrophotometer (Thermo Fisher Scientific, USA) and the 260/280 nm absorbance ratio was evaluated. In addition, to eliminate any potential genomic DNA contamination, the total RNA samples were treated with DNase I (Fermentas, USA). For gene expression analysis, one microgram of total RNA from each sample was reverse-transcribed using cDNA Synthesis Kit (TaKaRa, Japan) and examined employing SYBR Green Master Mix (TaKaRa, Japan) and specific primer sets synthesized by Macrogen Company, South Korea (Table 2). To test miR-141 and miR-200a expression, 100 ng total RNA from each sample was performed using a miR-CURY LNA Universal RT microRNA PCR kit (Exiqon, Denmark). Relative expression of mRNAs and microRNAs was evaluated by the 2−ΔΔCt method on ABI PRISM 7500 instrument (Applied Biosystems, USA) and normalized to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 small nuclear RNA as internal references respectively.
Table 2
|
Gene Name
|
Forward/Reverse
|
Primer Sequence
|
Amplicon Size (bp)
|
|
RARB
|
Forward
Reverse
|
5´TACAAACCCTGCTTCGTC 3´
5´ACACAGTTCTTATCTCGGTGA 3´
|
141
|
|
IL17A
|
Forward
Reverse
|
5` CATTGGTGTCACTGCTACTGC 3`
5` AAGGTGAGGTGGATCGGTTG 3`
|
201
|
|
RORC
|
Forward
Reverse
|
5` TGCCAGAATGACCAGATTGTGCTT 3`
5` GAACAGCTCCATGCCACCGTA 3`
|
132
|
|
IL23R
|
Forward
Reverse
|
5` CACATGGAATTCTGGGCTAACAG 3`
5` AGCAAAGACGATCATTCCCAAT 3`
|
111
|
|
GAPDH
|
Forward
Reverse
|
5` CCACTCCTCCACCTTTGACG 3`
5` CCACCACCCTGTTGCTGTAG 3`
|
107
|
Plasmid Construction And Dual-luciferase Reporter Assay
To specify direct interaction between microRNAs and 3´UTR of predicted target gene, sequenced purified PCR products of microRNA precursor were digested with XbaI and HindIII and ligated into the pBud-EGFP vector (Promega, USA). Synthesized wild type (wt) and mutant (mut) target gene–3´UTR were also cloned into the psiCHECK2 vector (Promega, USA). Both vectors were then co-transfected into HEK293T cells using Lipofectamine 2000 reagent (Invitrogen, USA) following the instructions. Cells were harvested at 48 h post-transfection, assayed for the firefly and Renilla luciferase activity using the Dual-Luciferase Reporter Assay System (Promega, USA). Each experiment was repeated in triplicate and changes in Renilla luciferase activity were calculated relative to firefly luciferase enzyme activity (internal control).
Statistical analysis
The results are expressed as the means ± standard error of the mean (SEM) of three independent experiments and analyzed by and GraphPad Prism 8 software. Independent sample t tests were used to evaluate the differences between groups. A P value of 0.05 or less was considered significant.
{kind=link}
{kind=link}
{kind=link}