ABCC10 was associated with Oxaliplatin sensitivity and CRC malignancy.
The chemo-resistance of cancer cells primarily attributed to the over-expression of the ABC transporter superfamily. Among these ABC transporters, we paid a special interest in ABCC10 because of the following reasons: ① According to the analysis on the clinic CRC specimens and the TCGA database, we found that although there was no significant difference in the ABCC10 expression between the tumors and adjacent normal tissues (Fig. 1a and b), the CRC patients with higher ABCC10 were apt to have metastasis, recurrence and shorter survival time (Fig. 1c~f). ② The IC50 of Oxaliplatin in 5 CRC cells including Caco-2, RKO, HCT-116, LS174T and HT-29 was 56.6 ng/mL, 65.8 ng/mL, 23.9 ng/mL, 60.8 ng/mL and 69.8 ng/mL, respectively, which was positively correlated with the expression level of ABCC10 (Fig. 1g~j), indicating that the CRC cells with lower ABCC10 were more Oxaliplatin sensitive.
Knock down of ABCC10 sensitized CRC cells to Oxaliplatin.
In siABCC10 (100 nM) treated Caco-2, LS174T and RKO cells, Oxaliplatin IC50 was declined from 56.2 ng/mL to 15.1 ng/mL, 72.6 ng/mL to 33.0 ng/mL, and 40.6 ng/mL to 8.9 ng/mL, respectively (Fig. 2a). Caco-2 cells treated with siABCC10 or siControl were exposed to 80 ng/mL of Oxaliplatin for 24 h and the intracellular accumulation of Oxaliplatin was measured by UPLC-MS/MS. Cells treated with 100 nM and 200 nM of siABCC10 had significantly increased Oxaliplatin accumulation (179.4±8.8 pg/mL and 152.1±5.1 pg/mL, respectively) compared with siControls (77.0±6.5 pg/mL) and 50 nM of siABCC10 treated cells (60.6±3.9 pg/mL) (Fig. 2b).
The increase of intracellular Oxaliplatin could be due to a decrease in the efflux of Oxaliplatin and/or an increase in the uptake of Oxaliplatin. To investigate the exact reason, ABCC10 knock-down Caco-2 cells and controls were incubated with 80 ng/mL of Oxaliplatin for 24 h, followed by further incubation in medium without Oxaliplatin for 0, 30, 60 and 120 min. The remained intracellular Oxaliplatin in shABCC10 group was 5814.3±590.4 pg/mL, 3947.1±508.8 pg/mL, 3240.0±200.0 pg/mL and 1305.0±330.0 pg/mL at 0, 30, 60 and 120 min, respectively, which was significantly increased compared with that in shControl group (1985.7±461.5 pg/mL, 307.5±13.6 pg/mL, 315.0±33.3 pg/mL and 440.0±40.8 pg/mL at 0, 30, 60 and 120 min, respectively) (Fig. 2c), demonstrating that the increased accumulation of intracellular Oxaliplatin by ABCC10 down-regulation was fundamentally because of the reduced efflux of Oxaliplatin.
These results indicated that Oxaliplatin was a substrate of ABCC10 transporter and the highly expressed ABCC10 accelerated Oxaliplatin efflux. Down-regulation of ABCC10 could impede Oxaliplatin efflux, which increased the intracellular Oxaliplatin accumulation and conferred the chemo-sensitivity of CRC cells.
ERS/UPR was activated in CRC.
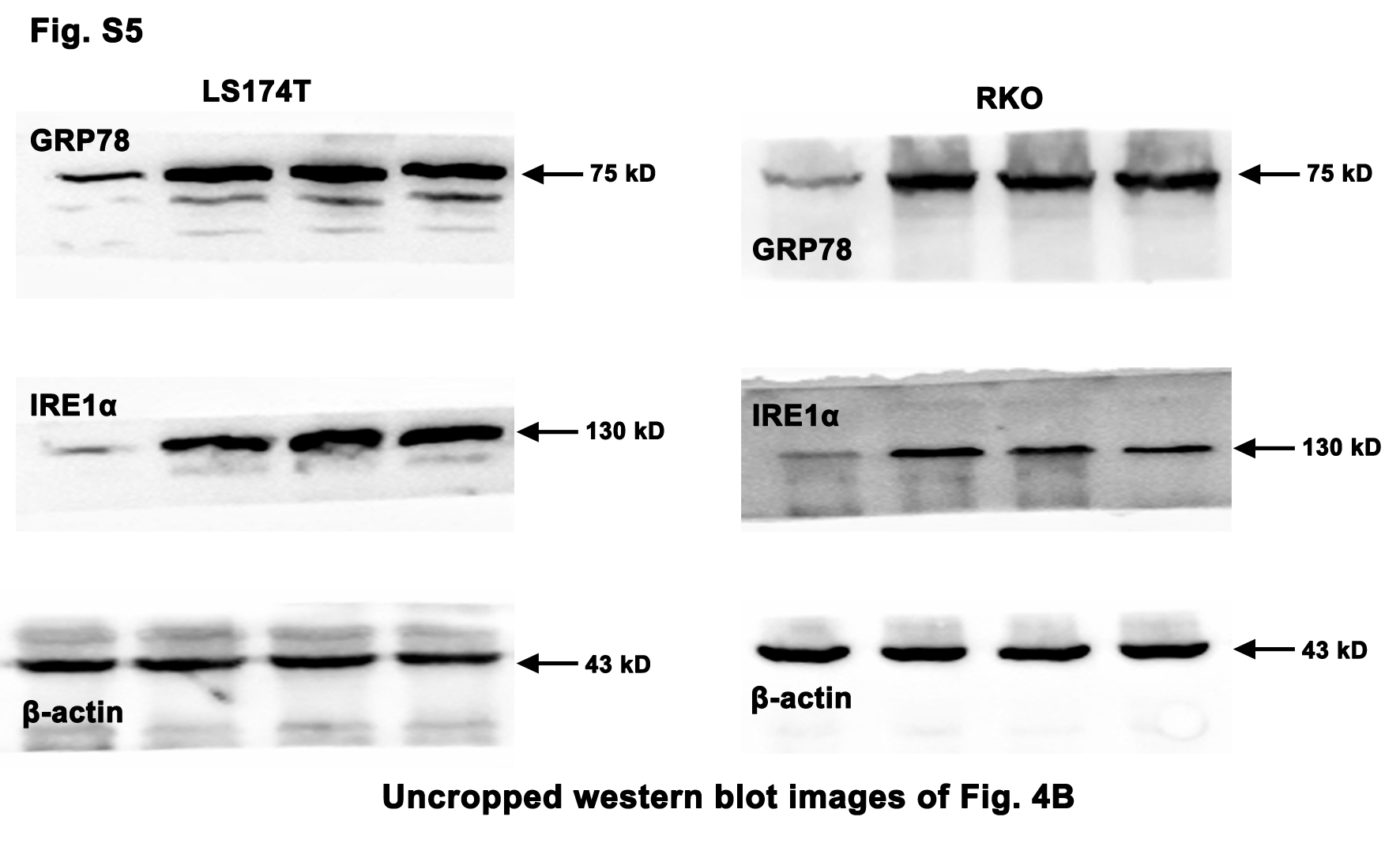
The ERS/UPR plays a dual role in tumor development, but its role in the Oxaliplatin resistance has not been fully elucidated. Here, the microarray and GO Enrichment analysis on CRC specimens revealed that the ERS/UPR, especially the IRE1α pathway, was activated in the CRC tissues compared with the adjacent normal tissues (Fig. 3a). GRP78, the biomarker of ERS, and its downstream IRE1α were hyper-expressed in tumors (Fig. 3b). The same results were obtained from the TCGA analysis showing that multiple genes in the IRE1α pathway were highly expressed in the CRC tissues (Fig. 3c).
Inactivation of ERS was unable to significantly reduce CRC cell viability.
In tumor cells, the adaptive ERS/UPR was conducive to the proliferation, invasion, angiogenesis and other malignant biological activities [19], prompting that inactivating ERS/UPR seems to be an anti-tumor approach. Therefore, in this study, the CRC cells were treated with an ERS inhibitor, 4-PBA (5 mM), for 12, 24 or 48 h. Unexpectedly, inactivation of ERS was incompetent to reduce CRC cell viability. Conversely, CRC cell viability was invigorated in some cases (Fig. 4a).
Mild ERS/UPR neither inhibited CRC cell viability.
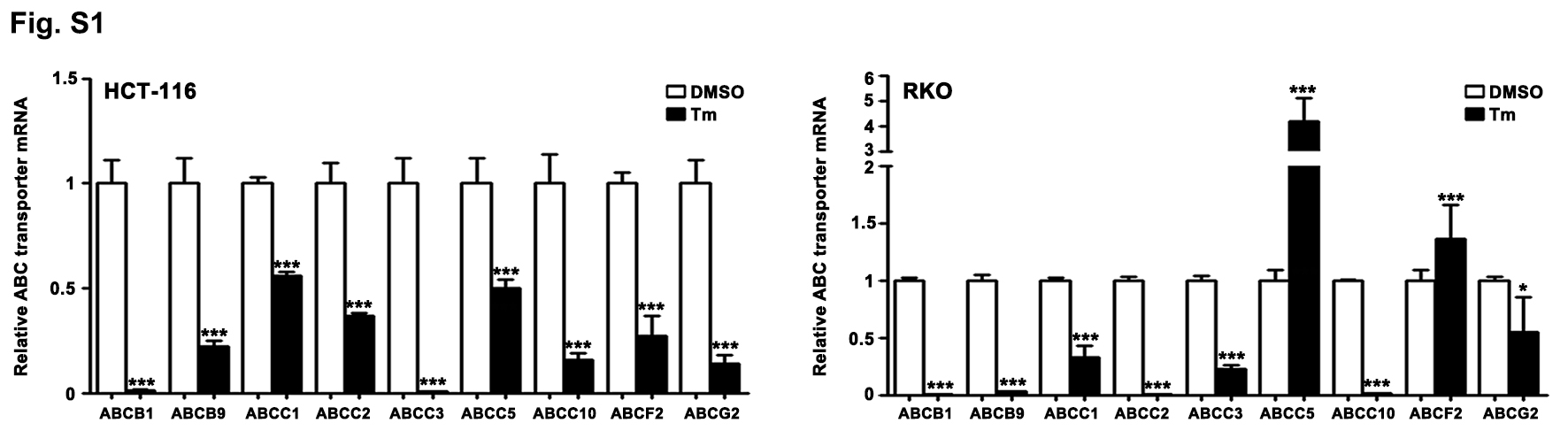
Given that the ERS/UPR could also exhibit anti-CRC effect, the CRC cells were exposed to different doses of Tm, which can provoke ERS by blocking the N-glycosylation in the post translational modification. As shown in Fig. 4, despite that low dose of Tm (0.5, 1 and 2 µg/mL) induced the ERS and the activation of IRE1α pathway in the CRC cells (Fig. 4b), the cell viability was not influenced significantly or just slightly reduced (Fig. 4c).
Intense ERS/UPR elicited significant anti-CRC effect.
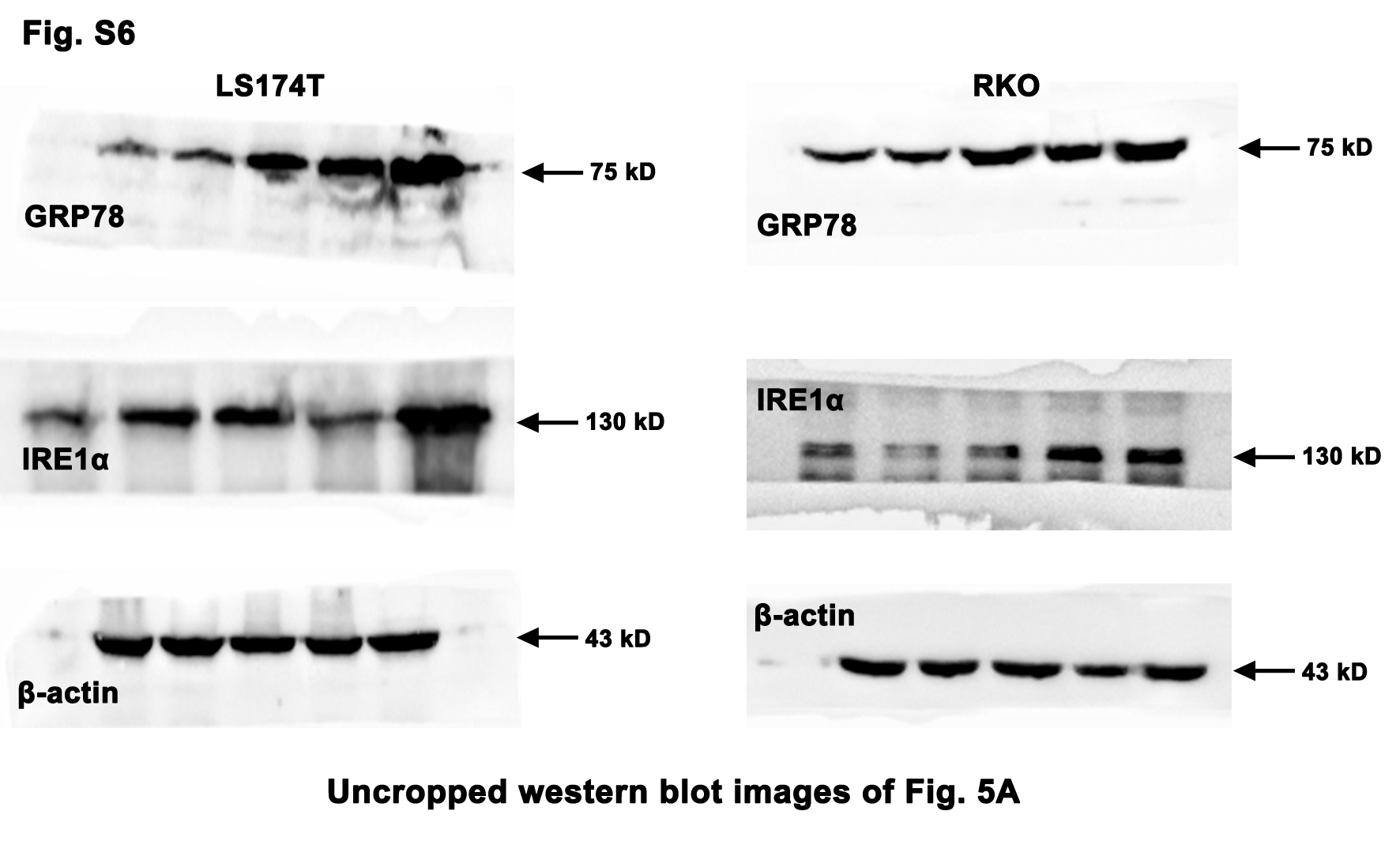
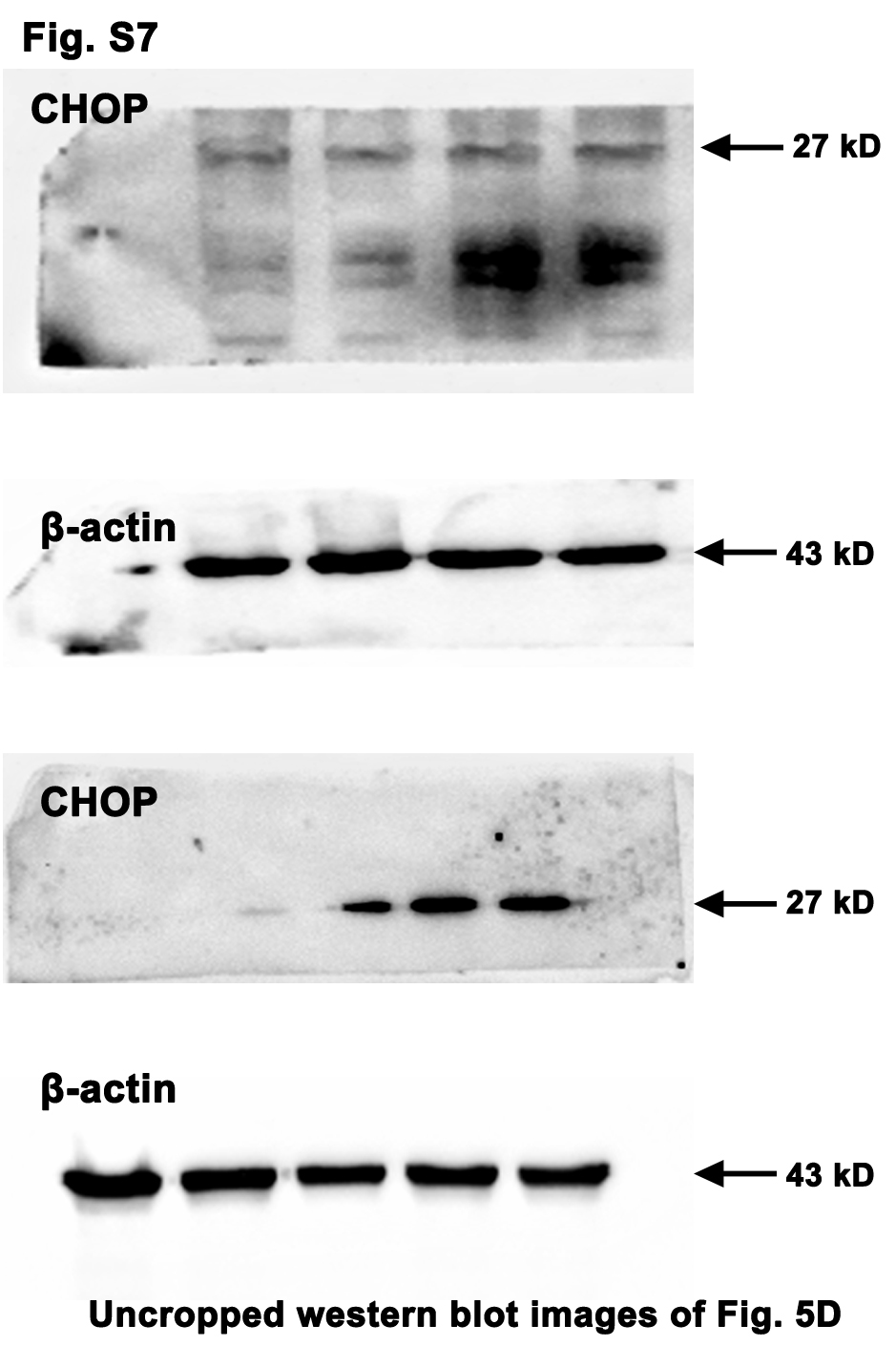
On the contrary, high dose of Tm (10 µg/mL) evidently abated the CRC cell viability upon the activation of the ERS/IRE1α pathway (Fig. 5a and b). It is worth noting that the viability of the NCM460 (normal colonic epithelial cell line) was not significantly decreased by high dose of Tm (Fig. 5c). Whereas, inhibition of the ERS by 4-PBA (5 mM) weakened its viability, which was not reversed by the additional Tm (Fig. 5c). Moreover, the intense ERS profoundly up-regulated the pro-apoptotic factor CHOP and promoted the CRC cell apoptosis, while the mild ERS did not (Fig. 5d and e). These results suggested the intense ERS/UPR instead of the mild ERS/UPR could elicit significant anti-CRC effect without obvious side effects on normal colonic epithelial cells.
Intense ERS/UPR sensitized CRC cells to Oxaliplatin.
Next, we wandered if the intense ERS was capable of raising the chemo-sensitivity to Oxaliplatin. The CRC cells were treated with increasing doses of Oxaliplatin in the presence or absence of Tm (10 µg/mL). When treated with Oxaliplatin alone, the IC50 was 52.2 ng/mL in Caco-2 cells and 67.0 ng/mL in RKO cells (Fig. 5f). The cell viability was further attenuated in response to additional Tm treatment, indicated by the reduced IC50 (20.4 ng/mL in Caco-2 cells and 15.6 ng/mL in RKO cells) (Fig. 5f). A rescue assay demonstrated that when the IRE1α pathway was blocked by STF-083010 (200 µM), the cell viability was evidently recovered (IC50=80.8 ng/mL in Caco-2 cells and IC50=71.4 ng/mL in RKO cells) (Fig. 5f). These results suggested that the intense ERS/UPR was competent to enhance Oxaliplatin sensitivity of CRC cells via IRE1α pathway.
Intense ERS/UPR increased intracellular Oxaliplatin accumulation by down-regulating ABCC10 via IRE1α pathway.
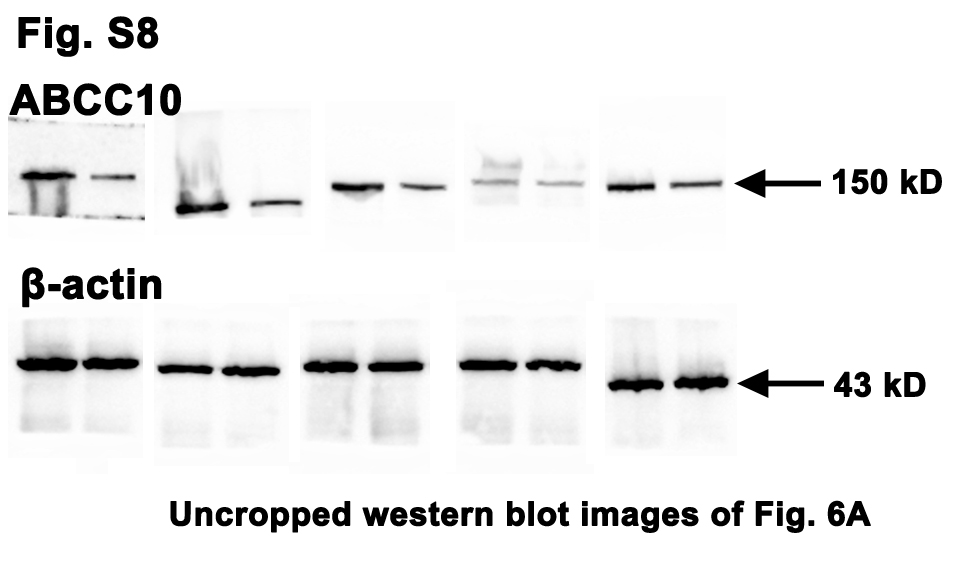
Since ABCC10 was responsible for Oxaliplatin efflux and sensitivity, we then asked whether ABCC10 was a target of the intense ERS/UPR. As shown in Fig. 6, high dose of Tm (10 µg/mL) significantly down-regulated ABCC10 protein in several CRC cells (Fig. 6a). However, when the IRE1α pathway was blocked by STF-083010 (200 µM), the expression of ABCC10 was restored (Fig. 6b). The intracellular accumulation of Oxaliplatin in Tm-treated CRC cells reached 6066.7±759.8 pg/mL, which was significantly increased compared with controls (1333.3±47.1 pg/mL). While, the blockage of IRE1α pathway by STF-083010 decreased Oxaliplatin accumulation to 8333.3±23.6 pg /mL (Fig. 6c). To confirm the disruptive role of Tm in ABCC10, a determined substrate of ABCC10, Paclitaxel [20], was used as a positive control in this study. Caco-2 cells were exposed to Paclitaxel (80 ng/mL) or Paclitaxel (80 ng/mL)+Tm (10 µg/mL) for 24 h. The intracellular accumulation of Paclitaxel was clearly increased in response to the intense ERS/UPR, but the increase was reversed by pre-treatment of STF-083010 (200 µM) (Fig. 6d).
Finally, we investigated the mechanism by which the intense ERS/UPR decreased ABCC10. Owing to the endoribonuclease domain, IRE1α mediates the cleavage of multiple RNAs in a process known as regulated IRE1-dependent decay (RIDD). The target mRNAs of RIDD contain a consensus sequence, CUGCAA, which could form a hairpin secondary structure in order to be cleaved. By the use of the RNAfold Web server, we found that ABCC10 mRNA (NM_001198934.2) contains the CUGCAA consensus sequence at the site of 1445~1450, suggesting that ABCC10 mRNA was a putative target of RIDD (Fig. 6e). Thus, we traced ABCC10 mRNA levels in Caco-2 cells exposed to Tm (10 µg/mL) at 2, 4 and 8 h, in the presence or the absence of the IRE1α RNase activity inhibitor STF-083010 (200 µM). ABCC10 mRNA was remarkably decreased when treated with Tm, while STF-083010 increased it up to 8 h (Fig. 6f).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}