Pulmonary samples
We carried out a retrospective review of mycobacterial cultures originating from Northern Tunisia (Tunis, Bizerte, and Zaghouan), which have been processed in the laboratory of mycobacteria of the Institut Pasteur de Tunis, Tunisia. From January 2002 to November 2016, a total of 10 671 pulmonary samples obtained from TB suspects have been processed (Fig. 1). After excluding follow up, recurrent, and relapse cases, the culture results of the remaining 10 466 specimens (7699 sputum, 2093 bronchial washes, 674 pleural fluids) were taken into account.
Except for a few cases, all processed pulmonary samples were from Tunisian patients who permanently resided in Tunisia. The vast majority of patients from Tunis lived in urban districts, whereas the majority of patients from the regions of Bizerte, and particularly those from Zaghouan, resided in rural areas. Virtually, all TB suspects from the latter two regions were admitted into their respective unique regional hospital, and hence, their forwarded specimens represented a full capture of the prevailing TB situation. By contrast, patients from Tunis, came from various healthcare sites from both the public and private sectors.
Mycobacterial isolation and phenotypic characterization
Briefly, pulmonary specimens (sputum, pleural fluid, bronchial wash) were processed by N-acetyl L cysteine-sodium hydroxide (NALC-NaOH) method, and inoculated into Löwenstein-Jensen (LJ) medium. Inoculated LJ slants were incubated at 37°C for eight weeks and examined every week.
Differentiation between MTBC and NTM colonies were initially performed by four standard biochemical tests: niacin, nitrate, heat-resistant catalase test (HRCT) and para- nitro benzoic acid (PNB). The NTMs were confirmed by PCR targeting the recA intein, as previously described [10], and were further characterized phenotypically by morphological character and biochemical tests, i.e colony morphology, growth rate, growth at various temperatures (25°C, 37°C and 44°C), pigment production in dark (schotochromogen), pigment production on exposure of light (photochromogen), absence of pigment production (non-chromogen), semi-quantitative Catalase test (SQCT), Thiophene2-carboxylic acid hydrazide (TCH) Susceptibility Test, Tween 80 hydrolysis, Aryl sulphatase test (3 days and 14 days), and β-Gal activities.
Data analysis
One isolate per patient was eligible for calculating PNTM isolation rate, which refers to the total number of PNTM divided by the total number of pulmonary specimens received in a year (annual rate) or during the study period (overall isolation rate). For patients with multiple PNTM isolations, we mostly took into consideration the very first isolate. PNTM isolation prevalence for the study period, or for a given year, was calculated as the total number of NTM positive cultures in a particular region divided by the population estimate and expressed per 100,000 population. In this study, we used the 2014 estimates of the Tunisian population, the most recent official estimates available: Tunis (2 643 695 inhabitants), Bizerte (568 219 inhabitants), and Zaghouan (176 945 inhabitants).
PCR amplification and nucleotide sequencing
From a few colonies cultivated on LJ slants, the DNA was extracted by thermolysis. For this purpose, the colonies are suspended in 100 μl of 1% EDTA / Triton Tris (TE / Triton) and inactivated at 80°C for 30 minutes.
PCR amplifications and sequencing of the rpoB, 16 S rRNA, hsp65, and sodA gene sequences were performed using the primer pairs listed in additional file 1.
PCR reactions consisted of a15-min inactivation period followed by 35 cycles of 95 °C for 30s, 60 °C for 1min (64°C for 1 min for rpoB, 52°C for 30 sec for 16S rRNA) and 72 °C for 2 min, with a final extension step at 72 °C for 5-7 min.
PCR amplifications were performed in a 50-μl PCR mixture containing 5 μl 10x buffer (Qiagen, Courtaboeuf, France), 200 mM each dNTP, 1.5 mM MgCl2, 1.25 U HotStarTaq polymerase (Qiagen), 1 μl each primer (10 pM), 33 μl nuclease-free water and 5 μl DNA template. Negative controls consisting of PCR mixture without DNA template were included in each PCR run. The PCR products were visualized by gel electrophoresis, treated with Shrimp Alkaline Phosphatase (SAP) and Exonuclease I (ExoI) (Sigma-Aldrich,USA), and sequenced in both directions using the BigDye Terminator sequencing kit (Applied Biosystems, Villebon-sur-Yvette, France) with an ABI PRISM 3100 automatic sequencer (ABI, USA). The sequences generated in the present study were deposited to the GeneBank. Their accession numbers are provided in additional file 2.
Nucleotide sequences were processed using BioEdit (BioEdit software, version 7.0.5), and aligned with the GenBank database (NCBI) using the Basic Local Alignment Search Tool (BLAST).
Species identification
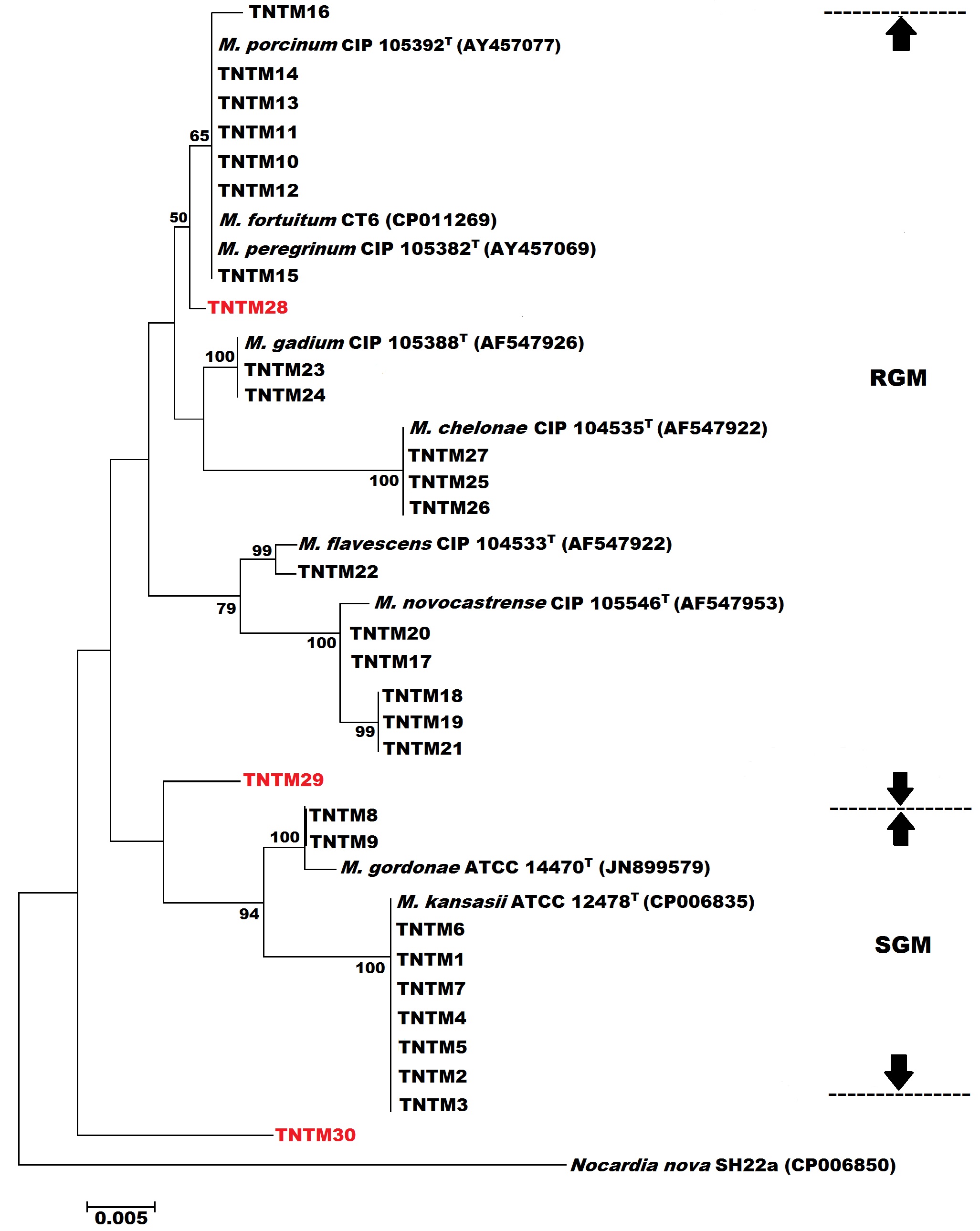
Species identification was primarily based on similarity rates with rpoB gene sequence of reference strains (http://www.bacterio.net/mycobacterium.html) [11]. Briefly, assignment of an isolate to a particular species was allowed if its rpoB sequence displayed at least 99.3% similarity to type strain for slow growers and 98.3% for rapidly growing mycobacteria. Isolates showing similarity rates below these thresholds but ≥ 94% were identified to complex level. Isolates displaying less than 94% similarity were referred to as rpoB unidentified Mycobacterium species (rUMS). With regard to 16S rRNA gene sequencing, identification was based on 100% matches.
Subtyping of Mycobacterium kansasii was performed by PCR-restriction fragment length polymorphism (PCR-RFLP) analysis of the hsp65 gene as described previously [12].
Phylogenetic analyses
Phylogenetic analyses were carried out with MEGA (Molecular Evolutionary Genetics Analysis, version 7.0) [13]. Phylogenetic tree analyses were performed using the neighbor-joining method based on the Kimura two-parameter model with 1000 bootstrap replicates. Nocardia nova was used as the outgroup.
{kind=link}