Plant materials, Genome sequencing and assembly
Gynura bicolor, a cultivated plant, voucher specimen (510918-1), was collected from Nanjing Botanical Garden Mem. Sun Yet-Sen. Gynura divaricata, a cultivated plant, voucher specimen (510918-6), was collected from Nanjing Botanical Garden Mem. Sun Yet-Sen. Gynura formosana, a cultivated plant, voucher specimen (512019-3), was collected from Kunming Botanical Garden. Gynura pseudochina, a wild plant, voucher specimen (512019-8), was collected from Wenshan Zhuang and Miao Autonomous Prefecture, Yunnan Province. All the plants were collected by Prof. Bingru Ren, and the specimens were deposited in the Herbarium of Institute of Botany, Jiangsu Province and Chinese Academy of Sciences.
The Gynura bicolor, G. divaricata, G. formosana and G. pseudochina plants were grown in a greenhouse with normal sunlight and temperature. The DNA was extracted from their fresh leaves by the CTAB method [17], and DNA degradation and contamination were monitored on 1% agarose gels.
Approximately 1.5 μg of the DNA sample was fragmented by sonication to a size of 350 bp. Then, the DNA fragments were end polished, poly A-tailed, and ligated with a full-length adaptor for Illumina sequencing, with further PCR amplification. After PCR product purification (AMPure XP system), libraries were analysed for size distribution by an Agilent 2100 Bioanalyzer and quantified by using real-time PCR.
The libraries constructed above were sequenced by the Illumina HiSeq X Ten platform, and 150 bp paired-end reads (PE150) were generated with an insert size of approximately 350 bp. Quality control (QC) removed reads with ≥10% unidentified nucleotides (N), > 50% bases having a phred quality < 5 and > 10 nt aligned to the adaptor, allowing ≤10% mismatches.
The Perl script NOVOPlasty 2.7.2 [18] was used to assemble the chloroplast genome sequence with a 50 K-mer. The chloroplast genome sequence of Dendrosenecio cheranganiensis (tribe Senecioneae) was selected as the reference genome. The family Asteraceae plant sequences used in the study were downloaded from GenBank as follows: Dendrosenecio brassiciformis (NC_037960.1), Dendrosenecio cheranganiensis (NC_037956.1), Dendrosenecio johnstonii (NC_037959.1), Dendrosenecio kilimanjari (NC_037957.1), Dendrosenecio meruensis (NC_037958.1), Jacobaea vulgaris (NC_037957.1), Ligularia hodgsonii (NC_039381.1), Ligularia intermedia (NC_039382.1), Ligularia jaluensis (NC_039383.1), Ligularia mongolica (NC_039384.1), Ligularia veitchiana (NC_039385.1), Artemisia gmelinii (NC_031399.1), and Chrysanthemum boreale (NC_037388.1).
Chloroplast genome annotation
The whole chloroplast genome sequences were annotated by Dual Organellar Genome Annotator [19] and GeSeq [20] with default parameters. Chloroplast genome sequences of tribe Senecioneae plants Dendrosenecio cheranganiensis and Pericallis hybrida were used as reference sequences. Subsequently, all tRNAs were verified by ARAGORN v1.2.38 [21] and tRNAscan-SE v2.0 [22]. A schematic diagram of the chloroplast genome with annotations was obtained by OGDRAW [23].
Repeat structure analysis
The microsatellite regions are a tract of repetitive DNA in which certain DNA motifs (ranging in length from 1-6 or more base pairs) are repeated, typically 5-50 times [24-25]. The Perl script Microsatellite identification tool (MISA, http://pgrc.ipk-gatersleben.de/misa/misa.html) was used to find the microsatellite regions of the chloroplast genome. Considering the features of plant chloroplasts, the numbers of each unit of continuous DNA motifs was set to 1-6, and the minus DNA motifs of each unit was 1-10, 2-6, 3-5, 4-5, 5-5, and 6-5. Forward, reverse, complement and palindromic repeat types were detected by the online tool REPuter [26]. The Hamming distance was set as 1, and the minimum repeat size was 30 bp.
Chloroplast genome analysis
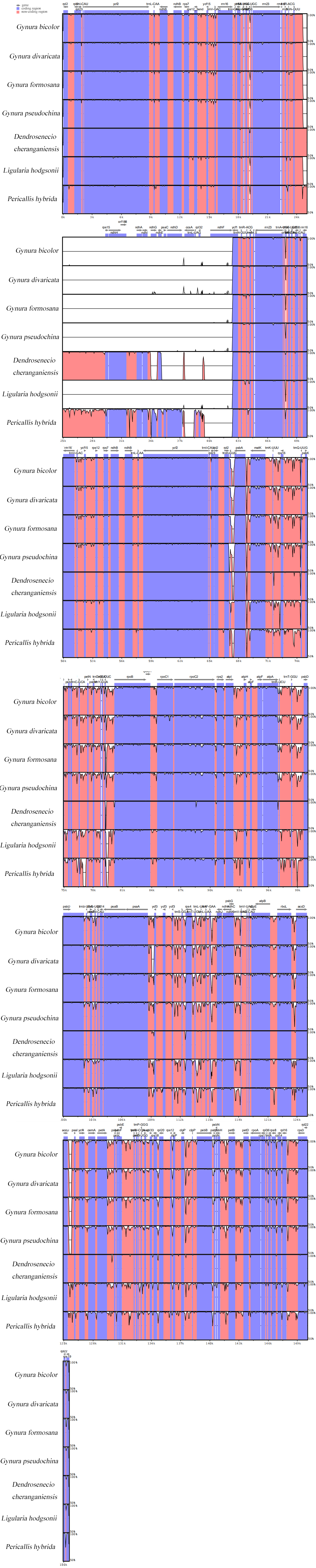
All the chloroplast genome sequences were aligned by MAFFT7.427 [27] on the FFT-NS-2 module. The different chloroplast genome sequences (LSC, SSC, IRa and IRb) concatenated together to make one sequence per species. Alignments of 7 selected genome sequences were visualized by mVISTA [28]. DNA polymorphism (nucleotide diversity) was calculated by DnaSPv5 [29] based on alignment results.
Molecular evolutionary rates (ω) between orthologous genes were estimated by calculating the ratio of the non-synonymous (dN)/synonymous substitution (dS) rates. Coding gene sequences of selected regions were extracted by using Artemis [30]. Gene sequences of each species were aligned by Clustal X [31] with default parameters, and the alignment results (dnd format) were converted to PML format by DAMBE [32] for subsequent analysis. The dN/dS value was calculated by the codeml module (seqtype=1, model=0, Nsites=1,7,8) in PAML4.9i [33]. Significant differences were calculated by the likelihood ratio test.
Phylogenetic analysis
The 16 chloroplast genome sequences of the tribe Senecioneae (family Asteraceae) were aligned by MAFFT, and the results were used to analyse the phylogenetic relationships. RAxML8 [34] was used to build a maximum likelihood tree with the GTRGAMMAI module and 1000 bootstrap replicates. Mrbayes3.2.7a [35] was used to build a Bayesian inference tree. The parameter settings were as follows: nst=6, rates=invgamma, burnin=500, Ngen=20000, Samplefreq=10, and Printfreq=100. Both the results of the ML tree and BI tree were visualized by FigTree V1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Divergence time estimation
The divergence time of 16 species was estimated by BEAST2 [36]. The oldest Artemisia fossil pollen has been recorded from the Eocene–Oligocene boundary [37-38]. The Asteraceae family plants Artemisia gmelinii and Chrysanthemum boreale were selected as the outgroup, and the node Artemisia–Chrysanthemum was constrained by using a lognormal distribution with an offset of 31 Ma and a mean and standard deviation of 0.5 [39]. The HKY nucleotide substitution model and the prior tree Yule model were selected with a strict clock. Each MCMC run had a chain length of 100,000,000 with sampling every 10,000 steps. Tracer [40] was used to read the ESS and trace value of logged statistics to access the results. Then, the divergence time was accessed by the Treeannotator program of BEAST2. The detailed settings were as follows: burnin percentage=50, posterior probability limit=0.0, target tree type=maximum clade credibility tree, and node heights=mean heights.
{kind=link}
{kind=link}
{kind=link}