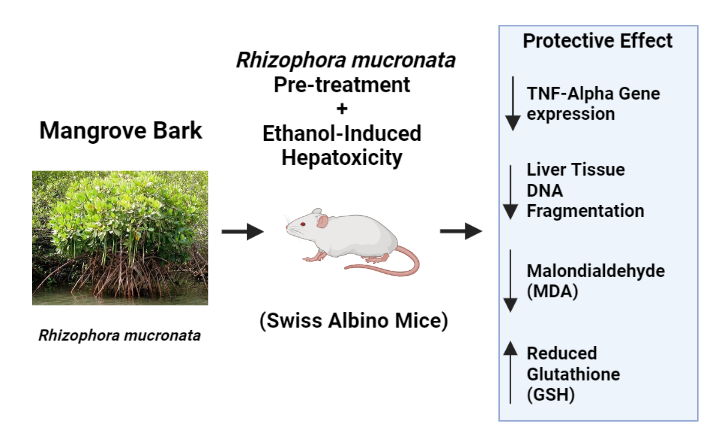
Liver and kidneys are both vital organs that are crucial for xenobiotic and drug elimination from our body (Galicia-Moreno et al., 2016). In contrast to the kidneys, the liver tissue, consisted of a mass of cells tunneled through with bile ducts and blood vessels, is mainly involved in the detoxification of viral infection, prolonged drug therapy, various toxicants (i.e., CCl4 and environmental pollutants, industrial chemicals, etc.), and chronic alcoholism (Zima et al., 2001; Ndoe et al., 2015; Unsal et al., 2020). Their metabolism and detoxification can generate a myriad of oxidative stress-related intermediate and end-products leading to hepatotoxicity, characterized by the hepatocyte death, capable of causing liver damage, liver injury and eventually liver diseases (Li et al., 2015). These harmful free radicals and reactive oxygen species may impair the prominent hepatic enzymatic and non-enzymatic antioxidant systems, and thus decrease the detoxification capability of the liver (He et al, 2017). So far, conventional medical therapy of liver failure or liver diseases such as drug-based treatment or even post-transplantation medication is still not denuded from side effects, requesting the urgent needs to discover new plant and plant-based formulations as safe medication therapies. These last decades, numerous studies have reported hepatoprotective activities of various natural products extracted from plants (Jiménez-Arellanes et al., 2016), including silymarin reaching clinical trials (Gillessen and Schmidt, 2020). We previously evaluated the in vitro hepatoprotective activities of BERM based on the reduction of the cytotoxicity in HepG2 cell line exposed to the combined BERM and toxicants (i.e., CCl4, ethanol and paracetamol) treatment (Padmanabhan and Jangle, 2014). In this present study, we demonstrated the hepatoprotective activities of BERM in ethanol-intoxicated mice associated with a decrease of ethanol-induced MDA production, enhancement of ethanol-decreased GSH production, and with the concomitant reduction of ethanol-induced hepatotoxicity, revealed by the down-regulation of TNF-α gene expression level and by the quasi-disappearance of fibrosis and apoptotic hepatocytes.
Oxidative stress has been mainly associated with the pathological process of ethanol-induced liver injury (Phaniendra et al., 2015). The production of MDA, commonly known as oxidative stress marker and as a marker of lipid peroxidation, was considerably enhanced in the ethanol study group in comparison with the untreated control group. The MDA overproduction due to ethanol-induced liver damage aligned with previous research studies (Chang et al., 2021). Another study reported that oxidative stress in brain due to ethanol consumption also elevated MDA levels (Das et al., 2007).
A crucial non-enzymatic antioxidant pertaining to oxidative stress is GSH, which removes H2O2 radicals and reacts directly with certain ROS (e.g., the hydroxyl radical) and nullify its toxic effects. In the present study, the ethanol toxicity group exhibited decreased levels of GSH in comparison with the control and treated groups, resulted in reduced synthesis of GSH, as previously reported (Husain et al., 2001). Observed in rats subjected to alcohol and tobacco smoke exposure, the generation of oxidative stress was also stated to decrease GSH levels in liver (Ignatowicz et al., 2013), which agreed with our present findings.
Oxygen radicals generated by the ethanol intoxication-induced injury play an important role in the stimulation of inflammation through up-regulation of inflammatory cytokines such as TNF-α ( Gutierrez-Ruiz et al., 2001). TNF-α is a key pro-inflammatory cytokine which induces the secretion of enzymes and other cytokines in various cells and tissues. In this present study, the prolonged exposure to ethanol leads to increased level of TNF-α gene in the toxic study group. Similar results were obtained by Nowak and Relja who demonstrated that NF-κB signaling pathway was activated during alcoholic liver disease, which resulted in the increased of gene expression levels of pro-inflammatory cytokines and chemokines (Nowak and Relja, 2020). In humans, chronic alcohol consumption is associated with increase in the production of serum pro-inflammatory cytokines (e.g., TNF-α, IL-1, IL-6, IL-8) (McClain and Cohen, 1989, McClain et al., 1999). Thus, the correlation between oxidative stress and inflammation within the course of alcoholic liver injury is indisputable. Moreover, improper metabolism of ROS ends up in the expression of hypoxia-inducible factor-1 alpha that may also increase TNF-α secretion, resulting in associate immune reaction that intensifies the liver injury (Wilson et al., 2014). In addition to play a major role in inflammation, TNF-α bound to its receptor, which initiates programmed death pathways such as apoptosis through activation of downstream kinases and proteases, including caspases (Fouad et al., 2019). A deeper investigation of the reverse effect of ethanol intoxication inducing apoptosis-related molecular mechanisms, including caspase-dependent (extrinsic) and mitochondria-dependent (intrinsic) pathways, contributing to hepatoprotective activities of BERM would be of interest.
The TUNEL assay was carried out for the detection of apoptotic cells that undergo massive DNA fragmentation during the final stages of apoptosis. The DNA damage may be incurred due to ethanol-induced oxidative stress exposed to the hepatocytes causing production of ROS and of TNF-α-induced cell death, which in turn lead to hepatic injury (Wang et al., 2016). The current study showed that ethanol intoxication towards mouse hepatocytes increased the number of apoptotic cells that was determined using TUNEL assay and observed with light microscopy, which agrees with previous studies using human alcoholic hepatitis specimens (Zhao et al., 1997; Natori et al., 2001). However, in this present study when pre-treated with either silymarin or BERM, a considerable decrease in the number of ethanol-induced apoptotic cells was noticed, confirming the in vivo hepatoprotective effect of BERM from alcohol intoxication.
{kind=link}