BAMscale algorithm
Peak quantification
Peaks can be quantified with BAMscale’s cov function, which takes as input a BED file with peak coordinates, and one or multiple BAM files, outputting raw read counts, FPKM, TPM and library size normalized peak scores. Paired-end reads can be quantified in two main ways: 1) using each read as a single entity, or 2) counting read pairs as one fragment. Additionally, it is possible to count reads that follow either the strand direction of each peak in the BED file, or simply calculate forward or reverse reads only.
During peak quantification BAMscale by default first reads the entire BAM file(s) to count the number of aligned reads using the selected alignment filters to get the effective library size. This approach gives more accurate alignment statistics than using the BAM index file, which has information on number of aligned reads only, containing duplicate reads and low quality reads as well. After calculating the effective library size, BAMscale counts the number of overlapping reads with each coordinate in the BAM file, followed by FPKM [47], TPM [48] and library size (scaled to the smallest library) normalization.
Creating coverage tracks from sequencing data
To generate normalized coverage tracks, the BAMscale “scale” function first imports the coverage of every bin (changeable) of the genome, followed by either genome size scaling (based on the length of the genome), or read count scaling. During genome size scaling, the scaling factor is calculated by dividing the total number of aligned bases with the genome size, which is obtained from the header of the BAM file. In cases where the number of bases exceed the genome size, scaling will reduce the per-bin coverage, while increasing the coverage when the sequenced bases are less than the genome size. The advantage of this approach is that each sample can be scaled separately. Alternatively, it is possible to scale multiple samples based on the library size. In these cases, the number of aligned reads is calculated for each sample, and scaling is done by scaling each sample to the smallest in the set. A drawback of this approach is that all samples have to be processed in parallel, which increases memory requirements (~500Mb for each sample when the bin size is set to 5bp). Additionally, it is possible to supply a scaling factor for each sample, that will be used to adjust the coverages.
We implemented an RNA-seq compatible option for creating coverage tracks with a difference in binning strategy. In RNA-seq mode, at cases where two adjacent bases in one bin have a coverage difference above 4 reads, the resolution is automatically changed to single base resolution. This enables the accurate representation of exon-intron boundaries.
Additionally, we implemented a signal smoothing option for coverage tracks. When smoothening a signal, the number of adjacent bins can be specified, which will be used to calculate the final signal of each bin.
In cases where two files are specified, different operations can be performed, such as calculating the log2 ratio of bins, or subtracting the values of bins.
For ease of use, we implemented predetermined settings to analyze replication timing data and END-seq data. In case of the “reptime” operation, the log2 coverage of two bam files are calculated for 100bp bin sizes, with signal smoothening set to 500 bins. In case of END-seq, we can set the operation to “endseq”, which creates stranded coverage tracks, or “endseqr”, which negates the coverage track of the negative strand for ease of visualization.
OK-seq data and replication fork directionality
In case of OK-seq data first the Watson (forward) and Crick (reverse) strand coverages are calculated consecutively. After importing the strand specific coverages, the replication fork directionality (RFD) is calculated as:
See Formula 1 in the Supplemental Files.
Where denotes the Crick read counts, denotes the Watson read counts for the i-th bin (based on [21]).
Sequenced data
NS-seq and replication timing sequencing of K562 cell lines
To produce high coverage sequencing of replication origins for the K562 human bone marrow derived cell line, we performed nascent-strand sequencing and replication timing sequencing available at GEO (GSE131417).
K562 NS-seq sample preparation
Replication origins were mapped using the nascent-strand sequencing and abundance assay [22]. Briefly, DNA fractionation was performed using a 5-30% sucrose gradient to collect DNA fractions ranging from 0.5 to 2 kb. Five prime single-strand DNA ends were phosphorylated by T4 polynucleotide kinase (T4PK) (NEB, M0201S). After phenol/chloroform treatment to remove T4 PK, DNA was precipitated, resuspended and then treated with lambda-exonuclease (NEB, M0262S) to remove genomic DNA fragments that lacked the phosphorylated RNA primer. After RNase treatment and DNA purification (Qiagen PCR purification kit, 28004), single stranded nascent strands were random-primed using the Klenow and DNA Prime Labeling System (Invitrogen, 18187013). Double-stranded nascent DNA (1 μg) was sequenced using the Genome Analyzer II (Illumina).
K562 replication timing sample preparation
To get the pure G1 phase cells, 1 × 108 K562 cells were washed twice with cold PBS and fractionated by elutriation at each of the following flow speeds: 15 × 2, 16 × 2, 17 × 2, 18 × 2 19 × 2, 20 × 2 ml/min. Each fraction was stained by DAPI in PBS and confirmed by FACS. Genomic DNA from G1 phase and asynchronized K562 cells was extracted simultaneously according to manufacturer’s instructions (Qiagen, 69506).
Data analysis
We demonstrate the capabilities of BAMscale on a wide set of sequencing datasets, such as ATAC-seq [31], ChIP-seq [35], replication timing data and END-seq [39], OK-seq [39, 42], single-cell Repli-seq and BrdU-IP [44] and stranded RNA-seq [38]. The complete list of analyzed samples, genome version and tissues of origins used in this study are shown in Table S1.
Alignment of sequencing data
The next-generation sequencing data GEO/ENA and SRA ids, along with sample type, aligner (and version) and genome build summarizing 27 processed sequencing experiments can be found in Table S1. ATAC-seq, ChIP-seq, OK-seq, END-seq and NS-seq were aligned with the bwa mem aligner [49] (version 0.7.17) or dragen pipeline [50] in case of the K562 replication timing. RNA-seq data was aligned using the STAR aligner [36] two-pass mode. Alignment settings were based on “best recall at base and read level” as shown in supplementary table 37 of [51] to obtain the best alignments. Aligned (unsorted) reads were sorted using samtools [30] (version 1.8), followed by duplicate marking using picard-tools (version 2.9.2).
Peak calling and coverage track generation for ATAC-seq, ChIP-seq and NS-seq
Peaks were identified using MACS [46] peak caller (version 2.1.1.20160309), using the –nomodel setting, and FDR set to 0.01 to filter low quality peaks. ATAC-seq peaks were called using the narrow setting, while histone peaks were called using the broad peak setting of MACS. In case of NS-seq data, both broad and narrow peaks were called, the top 10% of narrow peaks were intersected with the broad peaks to retrieve the highest scoring regions. Called peaks for each cell line and condition were sorted and merged using BEDTools [28] (version 2.27.1). Peak quantification was performed with the “cov” function of BAMscale separately for each sequencing type.
Gene expression quantification and differential expression analysis from RNA-seq data
Raw read counts for each gene were calculated using the TPMcalculator program [52]. Differential expression analysis between wild-type and KO samples were calculated using DESeq2 [53], where, as suggested in the manual, genes with less than ten reads on average were removed from the analysis. Scaling factors for each sample were obtained using DESeq2 sizeFactors() function. Scaled coverages were created with BAMscale “scale” function with operation set to “strandrnaR” and bin size set to 15 bases, scaling set to “-k custom” and scaling factor set to the reciprocal estimated factors from DESeq2 for each sample.
OK-seq data
BigWig signal of aligned OK-seq reads were created with the BAMscale “scale” function, with the operation set to “rfd” (replication fork directionality), all other parameters were set to default.
END-seq data
The strand specific coverage tracks for END-seq data were created the BAMscale “scale” function, with the operation set to “endseqr”. Two coverage tracks are created, one with the forward strand, and a separate track for negative strand reads, where the score is negated.
Replication timing data
Replication timing log2 ratio coverage tracks were created with the BAMscale “scale” function, with the operation set to “reptime”. The first specified BAM file is the G1-phase specific sequencing data, the second BAM file was the asynchronous cell-cycle BAM file. Replication-timing segments were identified the “Replication_timing_segmenter.R” script developed in R, and deposited on github along the BAMscale code.
Single-cell (sc) Repli-seq
Replication timing log2 ratio coverage tracks for single-cell replication timing data [44] were created with the BAMscale “scale” function. The operation parameter was set to log2, and to reproduce the original analysis results, we set the bin size was set to 50kb, and signal smoothening to 4 (resulting in 400kb smoothening). In case of the standard CBA/MsM samples the “CBMS1_ESC_single_G1_01” (GSM2904978) sample was used as the G1 phase reference, and sample “CBMS1_Day7Diff_ESC_single_G1_01” (GSM2905031) was used as the G1 phase reference for the 7-day differentiated CBA/MsM samples.
BrdU-IP replication timing data
The log2 coverage of early and late S phase BrdU-IP sequencing were calculated using the BAMscale “scale” function. Similarly to the scRepli-seq data, the operation parameter was set to log2, and to reproduce the original analysis results, we set the bin size was set to 50kb, and signal smoothening to 4 (resulting in 400kb smoothening).
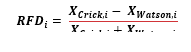{kind=link}