Mosquito rearing: Two strains of An. albimanus were used for this work: 1) An. albimanus Sanarate strain (Biosamples ID: SAMN10341946) (8), was used for gene characterization and quantification of basal gene expression. Rearing of this strain took place at the Universidad del Valle de Guatemala (UVG). Insectary conditions were 28 °C +/- 1 °C and relative humidity of 80% +/- 10%, with a photoperiod of 12:12 light-dark cycle. Colony larvae were fed daily a mixture of baby food Nestum® 5 cereals (Nestlé) and active yeast. Adults were fed ad libitum on a 10% sugar solution in sterile water and females were fed commercially available defibrinated sheep blood (Actividades Lucrativas, Guatemala, Guatemala) via a membrane feeder (Hemotek, United Kingdom) covered with Parafilm. 2) An. albimanus Santa Tecla (Stecla) strain (MRA-112 BioDefense Emerging Infections (BEI), Malaria Research and Reference Reagent Resource Center (MR4), Centers for Disease Control and Prevention (CDC), Atlanta, Georgia, USA) was used for RNAi experiments. Rearing conditions at the CDC were 27 °C, relative humidity of 80%, and a photoperiod of 12:12 light-dark cycle with a 30-minute dawn and dusk period. Colony larvae were fed Damien's diet (63) according to the MR4 rearing protocol (64). Adults were fed ad libitum on a 10% sugar solution with 0.2% methylparaben dissolved in sterile water, and females were fed commercially available defibrinated rabbit blood (Hemostat, Dixon, CA, USA) via a Parafilm covered glass feeding bell (Lily, Atlanta, GA, USA).
RNA extraction: For 3' RACE and PCR evaluation of the sex-biased actins expression, we extracted RNA from pools of: 1) 15 one-day-old virgin females, 2) 15 one-day-old virgin males, 3) five female pupae, 4) five male pupae, 5) seven L4 larvae, and 6) 40 L3 larvae of An. albimanus strain Sanarate. The number of individuals per pool was selected based on previous normalizations of RNA obtained per wet body weight. The extractions were performed using the SV total RNA isolation system kit (Promega, WI, USA), following the manufacturer's protocol. Briefly, mosquitoes were homogenized using a pellet pestle cordless motor and sterile pestles in 175 µl of lysis buffer and incubated in a cold rack for 20 min. 350 µl of RNA dilution buffer was added and mixed by inversion. This lysate was centrifuged at 13,000 rpm, 10 min at room temperature, the supernatant was removed and mixed with 200 µl 95% ethanol, transferred to spin column and centrifuged at 13,000 rpm for 1 min. The column was washed with 600 µl of RNA wash solution, and the sample was treated with DNAse incubation mix for 30 min, followed by the addition of 200 µl stop solution. The column was centrifuged at the same conditions as before and washed twice with RNA wash solution. RNA was eluted with 100 µl of RNase free water.
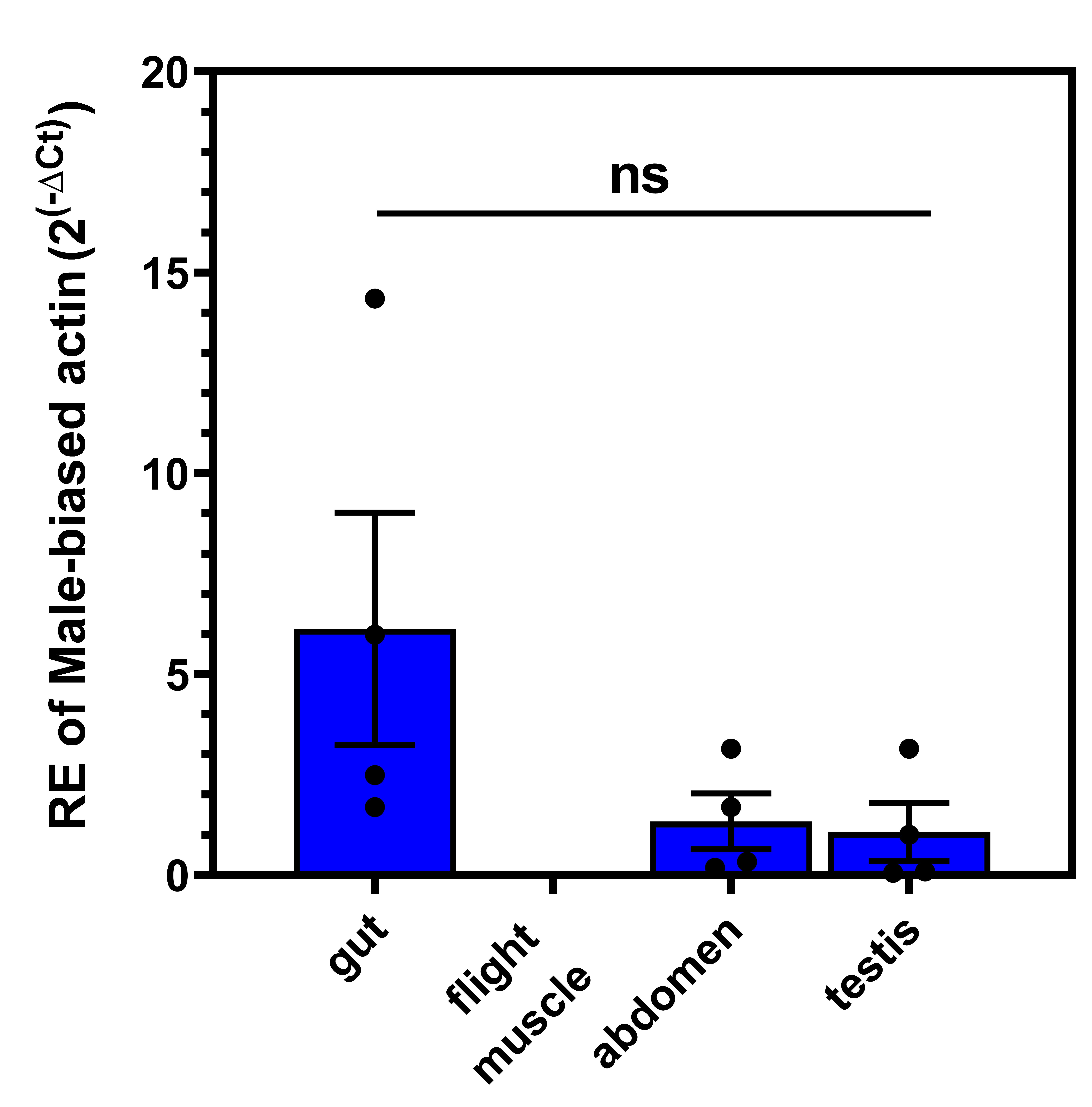
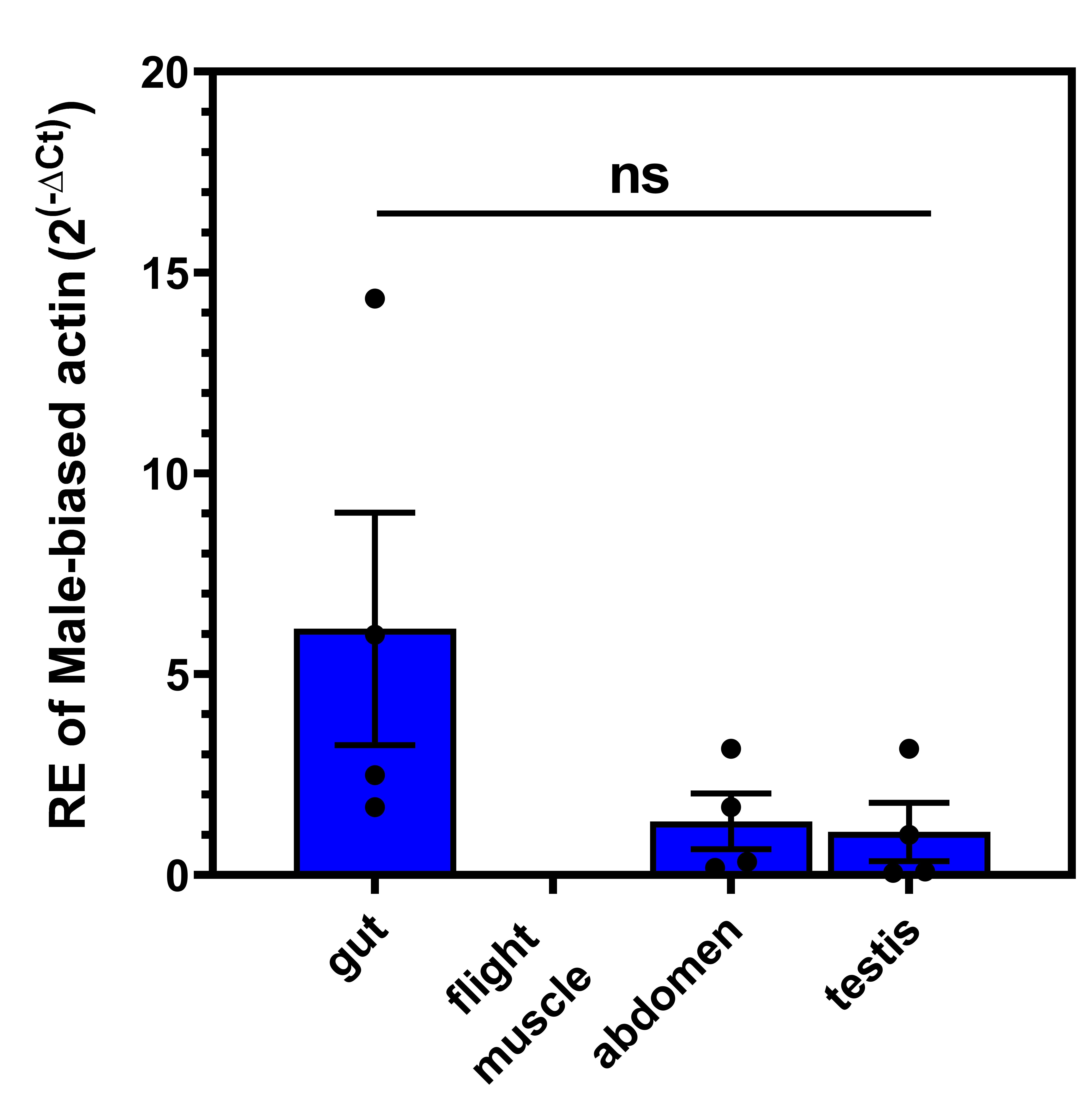
For the evaluation of female-biased actin expression in several adult tissues, two pools of five individuals per biological replica (n = 3) were dissected in ice-cold PBS. RNA extractions were performed using the RNeasy Micro kit (Qiagen, MD, USA) according to the manufacturer's protocol.
For cloning of the female-biased actin, RNA was extracted from a pool of eight adult female mosquitoes and five female pupae of An. albimanus Stecla strain using the RNeasy Mini Kit according to manufacturer´s protocol (Qiagen, MD, USA). Briefly, samples were homogenized using a pellet pestle cordless motor and sterile pestles in 350 µl of RLT buffer. After centrifugation, one volume of 70% ethanol was added to the lysate and transferred to the RNeasy Mini spin column. Two consecutive washes with 700 µl of RW1 buffer and one with 500 µl of RPE buffer were done before resuspending the RNA in 30 µl of RNase-free water. Quantification of RNA was done with a Nanodrop OneC (Thermo Fisher, MA, USA) and 1 µg of RNA was treated with 1.5 units of RQ1 DNase (Promega, WI, USA) for 1 h at 37 °C, followed by inactivation for 10 min at 65 °C.
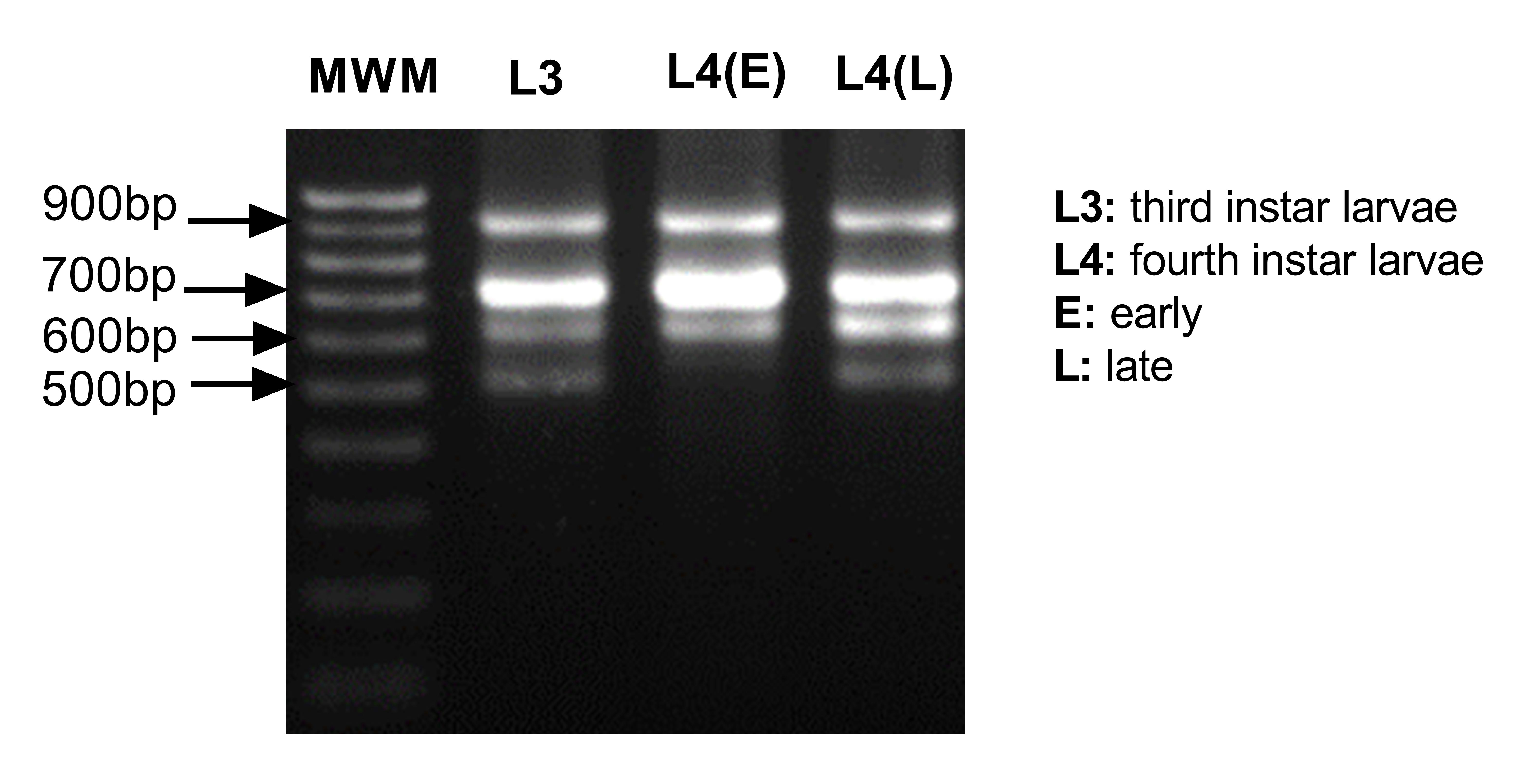
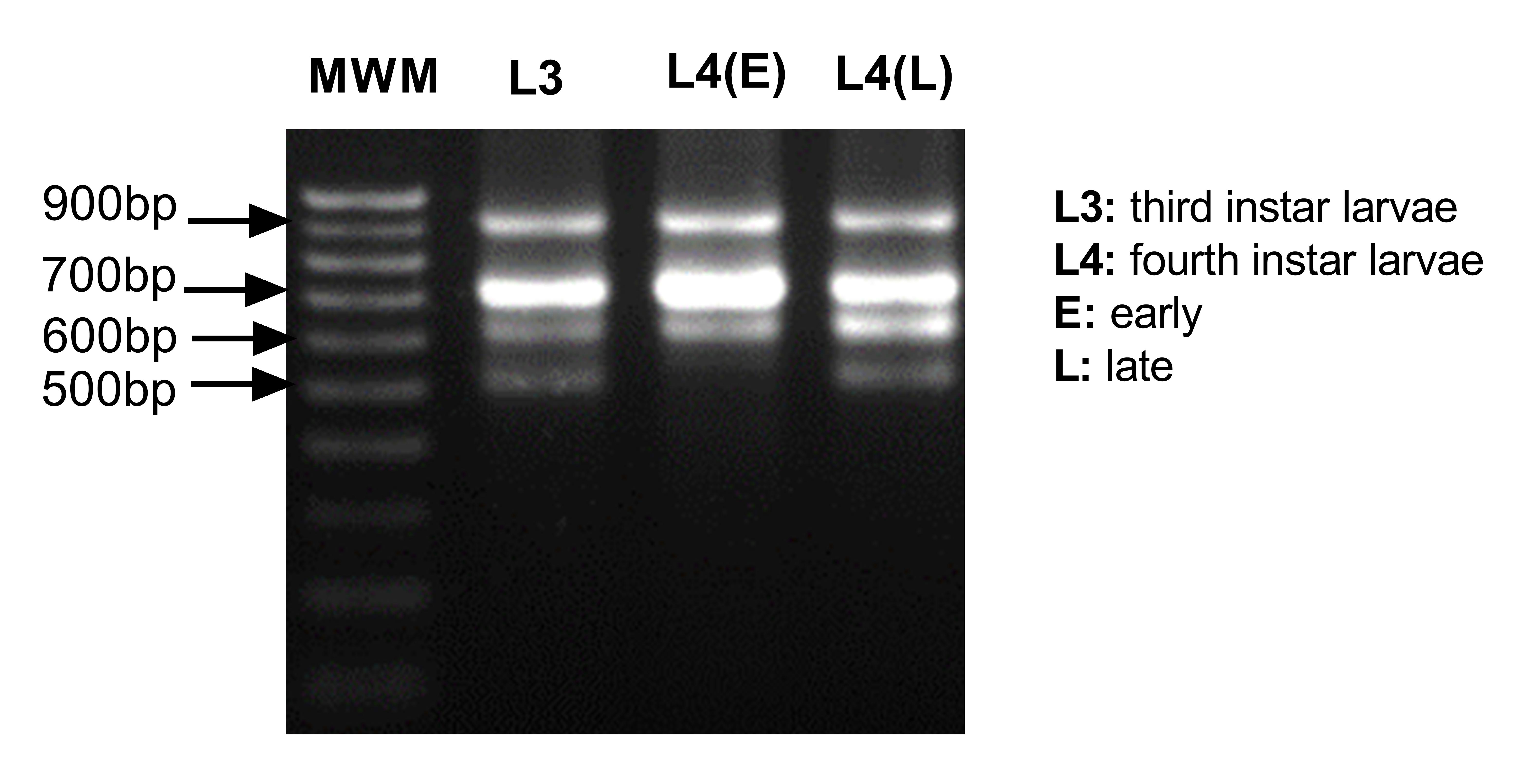
3' Rapid amplification of cDNA ends (RACE): We performed 3' RACE with the kit 3' RACE System for Rapid Amplification of cDNA Ends (Invitrogen, MA, USA). One µg of total RNA was used to generate the cDNA by the manufacturer's instructions. Then 1 µl of this cDNA was used to amplify the 3' UTR region using 0.4 µM of actin primer FMActRace_1F (5'- CGATCAAGATCAAGATCATTGCC-3') and 0.4 µM of UAP primer provided by the manufacturer in a 25 µl reaction using the GoTaq Hot Start colorless Master Mix (Promega). We performed the amplification as follows: 95 °C for 2 min, 30 cycles of 95 °C for 30 s, 61 °C for 30 s and 72 °C-1 min and a final extension of 72 °C for 5 min. We gel pricked the 600 bp and 500 bp bands that showed differential expression in females and males and amplified using the same conditions described earlier, with the only modification that 20 cycles of amplification were used. Both fragments were ligated to the pGEM-T easy vector (Promega) and transformed it into E. coli XL-1 blue. Clones of each fragment were sequenced and the sequences used to design female-biased and male-biased actin specific primers within the 3' UTR.
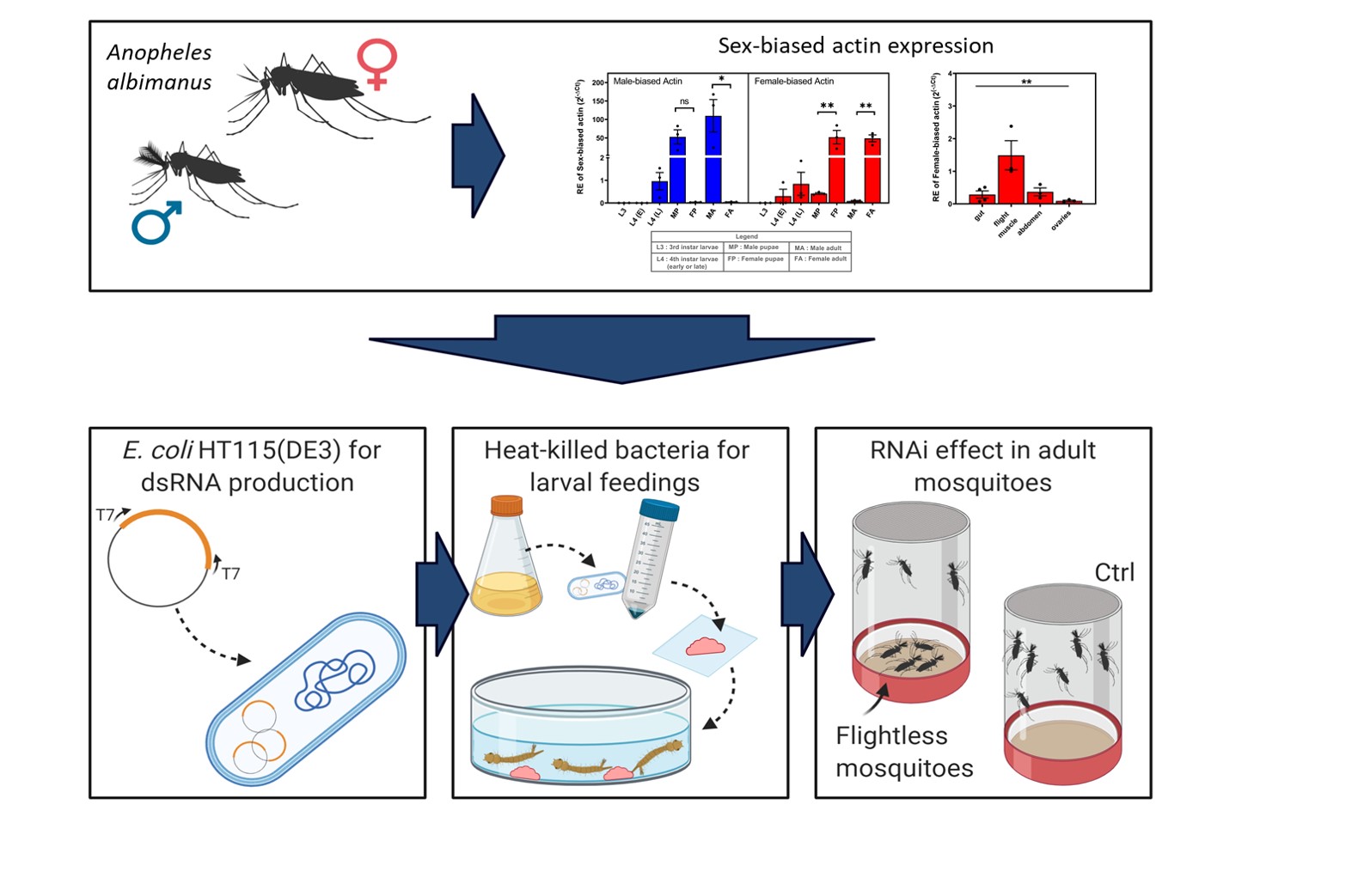
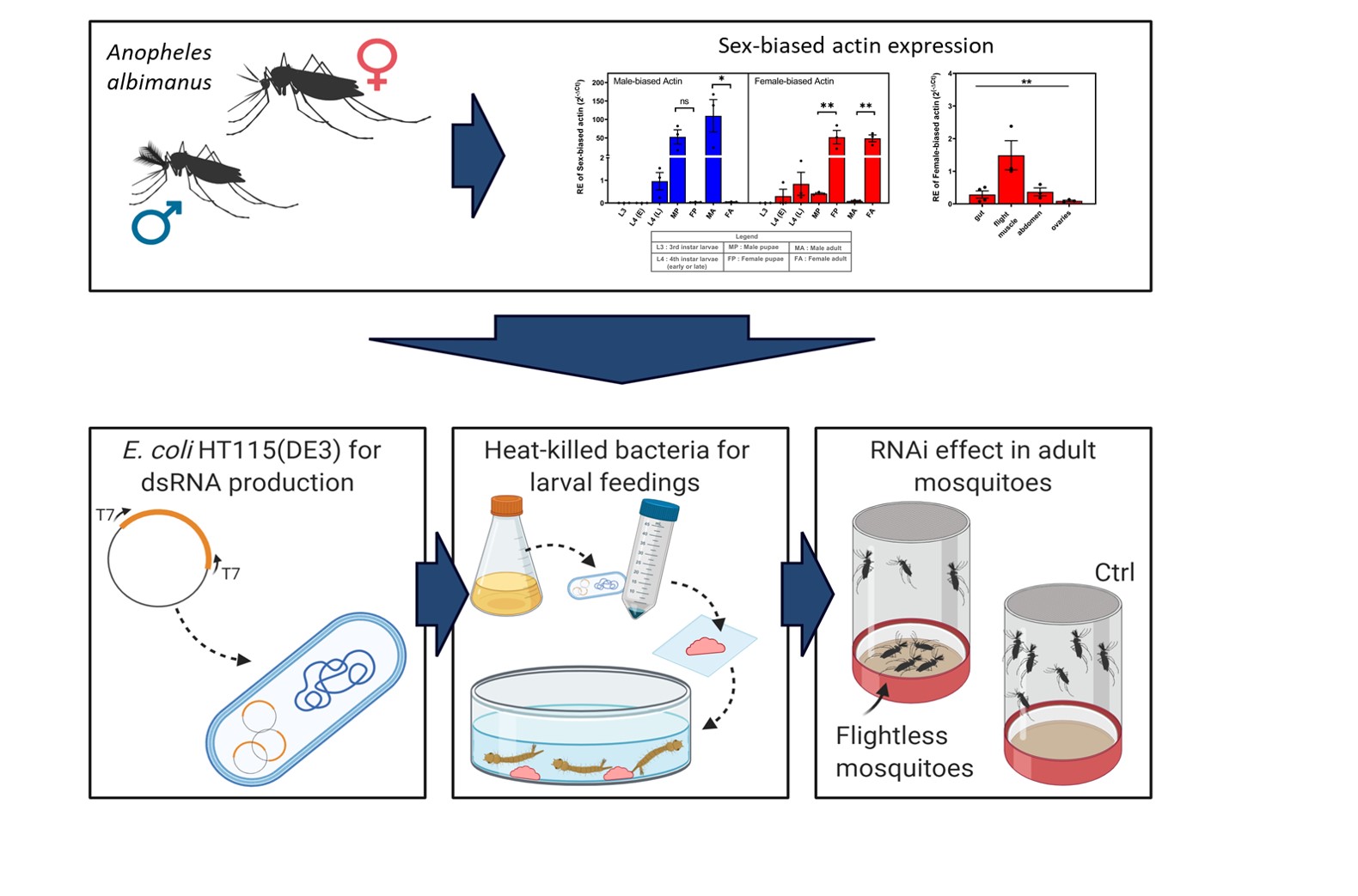
Sex-biased actin expression: Quantitative PCR was done to assess the expression level of the sex-biased actins during the life cycle and in the different tissues of the adults of An. albimanus mosquitoes. RNA extraction was performed as previously described. The cDNA was generated from 1 µg of total RNA using the GoScript Reverse Transcription Kit with a mixture of Random primers plus oligo(dT) (Promega) as per manufacturer's instructions. The following primers were used: qPCRfemaleUTR_2F (5´-GACATCCAATCACTACATCC-3´) and qPCRfemaleUTR_2R (5´-CAAAGGCAAACACAGCTAAC-3´) for the female-biased actin, and qPCR_Male3UTR_1F (5´-CCAACATCAACACCAACTTC-3´) and qPCR_Male3UTR_1R (5´-GTCGAAGAAGATTTGCACAG-3´) for the male-biased actin amplification. The RpS4 (AALB005332) and RpL49 (AALB008968) reference genes were amplified with the following primers: qPCR_RpS4_1F (5'-AGGTGATGGAGGTGCTGAAG-3´) with qPCR_RpS4_1R (5'- CGATGATGAACACGTTGGAG-3') and qPCR_RpL49_1F (5'-AGGGTGGAAGGTGATATCTG-3') with qPCR_RpL49_1R (5'- ACATGGTCGCCCTTAAATG-3'). To measure expression in each biological replicate of the different life-stages, qPCR was carried out with Power Up SYBR green master mix (Thermo Fisher) with 0.33 µM of each primer and 2.5 µl of cDNA diluted at 1:500 in a final volume of 10 µl. All samples were run in technical triplicates. Genes were amplified in the Lightcycler 96 (Roche, Switzerland) thermal cycler as follows: 50 °C for 60 s and 95 °C for 120 s, followed by 50 cycles of: 95 °C for 15 s and 60 °C for 60 s. Melting curves of 95 °C for 15 s, 60 °C for 60 s and 95 °C for 16 s were done after each run. Delta cycle threshold (∆Ct) method was performed using the average Cts of both Rps4 and Rps49 reference genes and performing the comparison of the relative expression to the L3 stage. Samples were analyzed using ANOVA test with Tukey's comparisons. To measure expression in each biological replicate of the different tissues, qPCR was prepared as before with the difference that the cDNA was diluted at 1:20. All samples were run in technical duplicate. PCR was run in a Quant Studio 6 (Applied Biosystems, CA, USA) thermal cycler as follow: 50 °C for 2 min, 95 °C for 10 min, and 50 cycles of 95 °C for 15 s and 60 °C for 60 s. Melting curves of 95 °C for 15 s, 60 °C for 60 s, and 95 °C for 15 sec were done for each run. Delta-delta Ct (∆∆Ct) analysis was performed using the average of Cts of both Ribosomal protein S7 (Rps7) and Actin housekeeping HK reference genes for analysis of transcript levels of pupa that receive actin A dsRNA and Rps7 only for pupae that were fed with Actin B and compared to pupae fed dsRNA of the ant gene as a control.
Female-biased actin cloning: Primers for two dsRNA were designed based on the sequenced regions. For 3´-UTR: AaACT3UTR_F: (5´-GCACAAATGATGGTGGCTAAAG-3´) and AaACT3UTR_R_T7: (5´-TAATACGACTCACTATAGGGCTCAAAGGCAAACACAGCTAAC-3´) (designated Actin A) and for coding region plus 3´-UTR: FMActRace_1F and AaACT3UTR_R_T7 (designated Actin B). The T7 promoter of the pGEM-T Easy vector and the T7 added in the reverse primers (underlined) were used to produce both strands of the dsRNA. Complementary DNA was produced using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher) with a mixture of random primers and oligo(dT). Conventional PCRs were performed with 0.4 µM of each primer and the AccuStart™ II GelTrack™ PCR SuperMix (Quantabio, MA, USA), using a 1:5 dilution of cDNA from female and male pupae and adults of An. albimanus as a template to corroborate the expression and the following program: 94 °C for 3 min, 30 cycles of 94 °C for 45 sec, 61 °C for 30 sec, and 72 °C for 30 sec, followed by a last step at 72 °C for 5 min. The obtained fragments were cloned into the pGEM-T-easy vector and then transformed into E. coli HT115 (DE3) for production of dsRNA. Transformation of E. coli HT115 (DE3) was performed as described by Timmons et al, 2001 (37).
Preparation of dsRNA: Pricks of fresh bacteria colonies were used for inoculum of overnight cultures. The bacteria used were either E. coli HT115(DE3)-Actin (Actin A or B) and E. coli HT115(DE3)-Ant, a non-related gene from Arabidopsis thaliana (GenBank: U41339.1), which is an APETALA2-like gene with pleiotropic roles in ovule development and floral organ growth, that we had previously cloned into E. coli HT115 (DE3) for control(65)). Colonies from Luria-Bertani (LB) agar plates with tetracycline (12.5 µg/ml) and ampicillin (100 µg/ml) were used as inoculum for each starting culture (10 ml). An overnight incubation in LB broth with tetracycline (12.5 µg/ml) and ampicillin (100 µg/ml) was done at 37 °C and 170 rpm. This culture was diluted 15:100 with 2xYT medium plus both antibiotics (same concentration as before) and incubated at 37 °C at 170 rpm until they reached OD600 = 0.4 (approximately 0.5 h). Once the culture reached OD600 = 0.4, the dsRNA production was induced with the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG), 0.4 mM final concentration, and a two-hour incubation at the same conditions (3.35 × 107 – 4.02 × 107 CFU/ml). This culture was centrifuged for 10 min at 4,000 rpm and 10 °C, and the cell pellet was washed twice in one volume of sodium phosphate buffer (PBS) (Thermo Fisher) and later re-suspended in half the initial volume in PBS. The E. coli cells were heat-killed by placing the tube with the resuspended pellet in boiling water for 15 min. The resuspended inactivated cells were aliquoted and stored at -70 °C.
dsRNA quantification
We used 3 aliquots of 250 µl of the inactivated cells to extract and quantify dsRNA. We centrifuged each tube of cells at 3,700 x g for 10 min at 4 °C, removed the supernatant, resuspended the pellet with 50 µl of 1% SDS in PBS and boiled them for 2 min in a water bath. After 10 min at room temperature, 64 µl of RNAse buffer (300 mM sodium acetate, 10 mM Tris-HCl pH 7.5 and 5 mM EDTA) and 2 µg of RNase A (Sigma Aldrich, MO, USA) were added and the mixture incubated at 37 °C for 5 min to degrade single stranded RNA. We extracted the dsRNA with 750 µl of Trizol LS Reagent (Invitrogen, MA, USA) according to manufacturer instructions. The RNA pellet was resuspended in 20 µl of DEPC water and 1 µl of the suspension was quantified on a Nanodrop one C (Thermo Fisher).
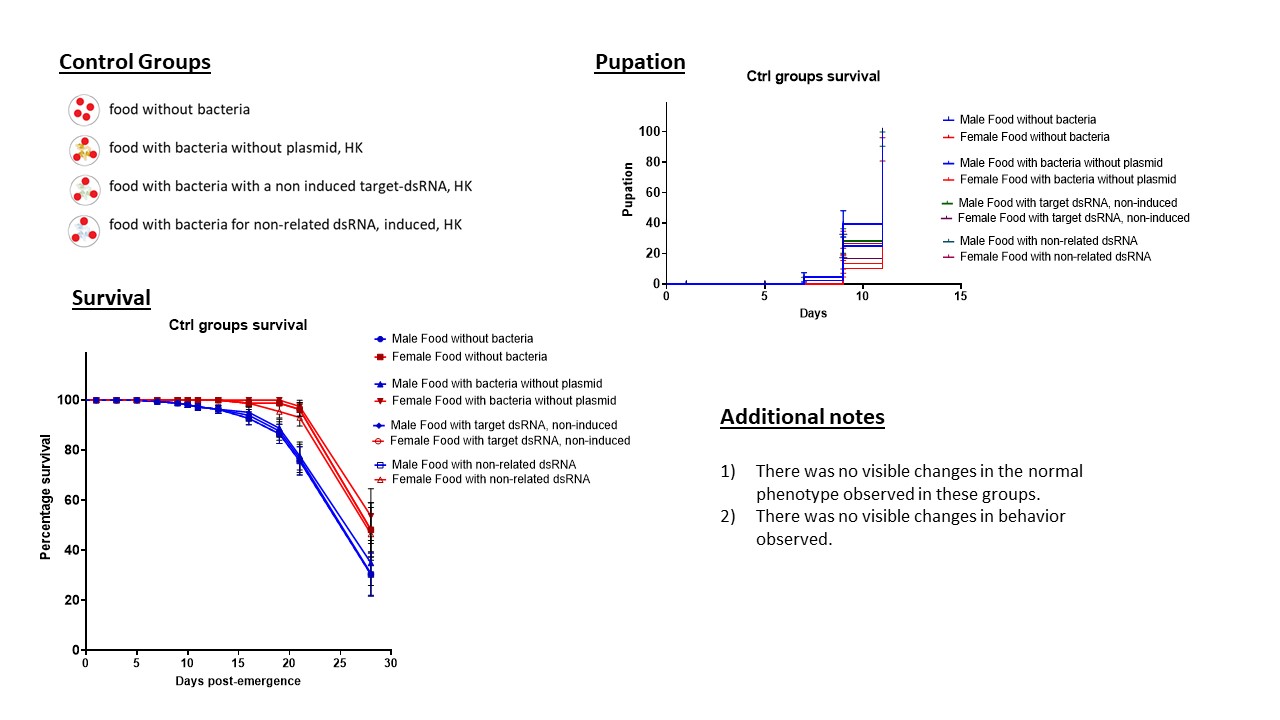
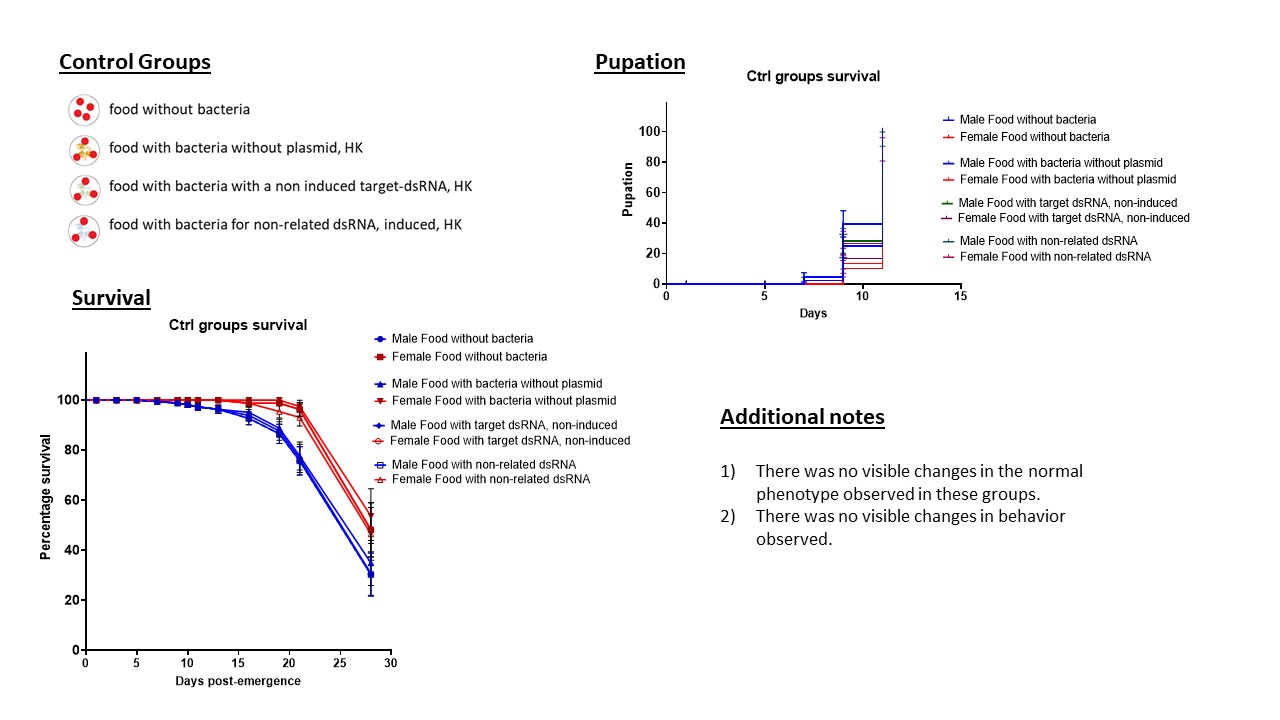
RNAi feeding experiments in larvae: Groups of 20 larvae (L2) of An. albimanus Stecla were set in individual 150 mm x 25 mm Petri dishes with 80 ml of water. Insectary conditions of temperature, humidity, and light were maintained as described in the mosquito rearing section. Feeding conditions were as following: an artificial bacterial diet (ABD), previously described by Taracena et al. 2019 (35), was added to the Petri dishes with the larvae, for a space of 4 h per day. Briefly, 200 µl of inactivated bacteria in PBS were complemented with 200 mg of a mixture of food (40% fish food (Goldfish, Tetra, Germany), 43% guar gum (Sigma-Aldrich) and 17% Active Yeast) and fed to each replica. After the 4 h, the dsRNA mixture was removed and we fed the larvae with conventional food (in this case, Baby 5 cereals Nestum® and active yeast). Negative controls for method validation were established in the following groups: 1) food without bacteria, 2) food with bacteria without plasmid for dsRNA production, 3) food with bacteria with a plasmid for the target dsRNA, but non induced, and 4) food with bacteria with non-related dsRNA. After the determination that no significant difference was observed amongst the negative controls (Additional File 6), the group of Ant-dsRNA was kept for reference in all experiments with actin genes. The larvae were fed daily with ABD until they reached pupation. Quantification of the dsRNA allowed to determine that the feedings were delivering an equivalent of approximately 5.75 µg of Ant-dsRNA, 11.90 µg for Actin A, and 9.0 µg for Actin B at the beginning of each feeding. Degradation of the dsRNA in the medium or inside the larvae was not measured. For each biological replicate (N = 80) we had four internal replicates, each with 20 larvae. Observed phenotypes were recorded for all experiments. T-test was performed in comparison to the control group for the flightless phenotype. The Log-Rank Mantel-Cox test was performed for survival curves in comparison to the control group.
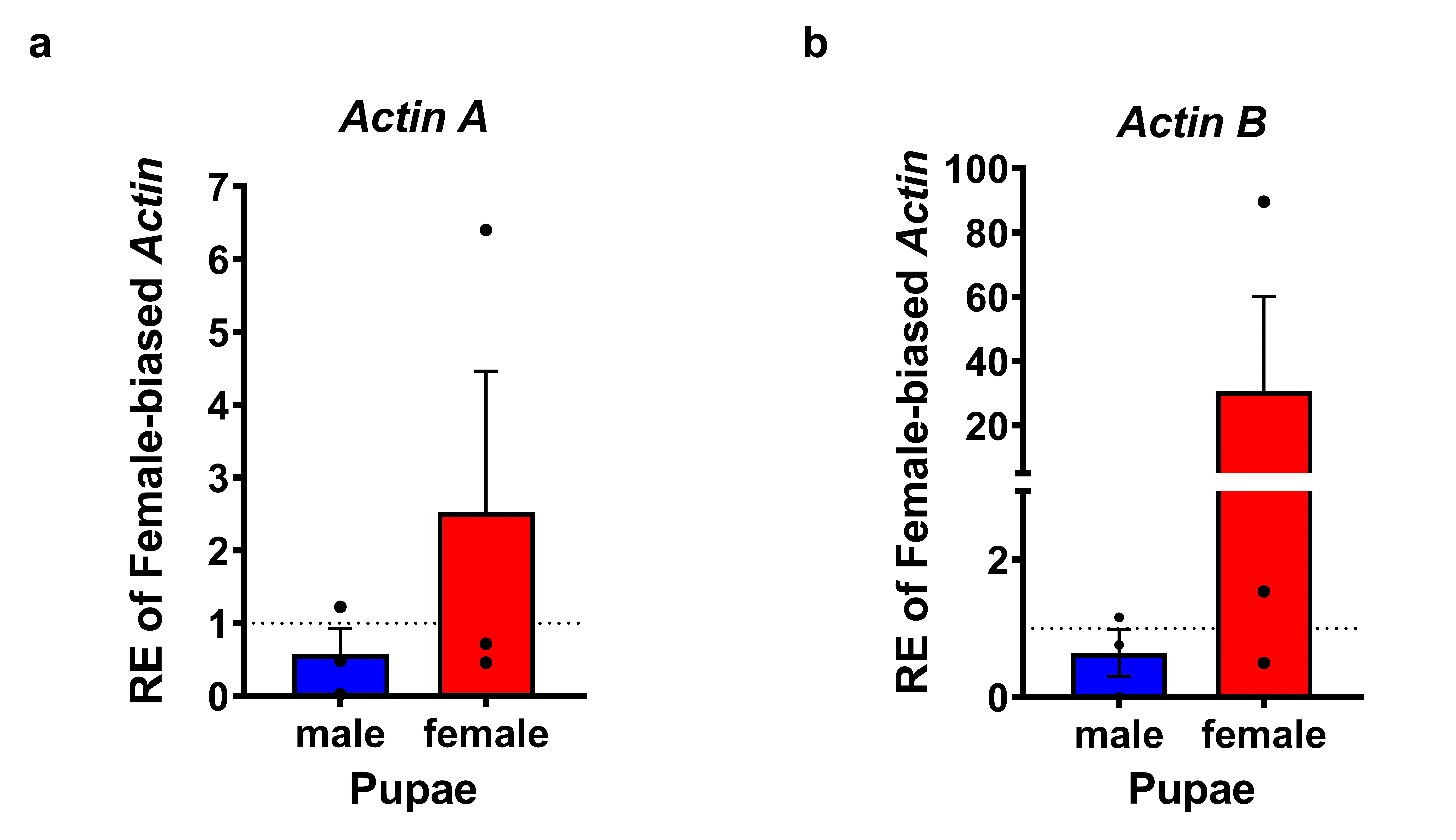
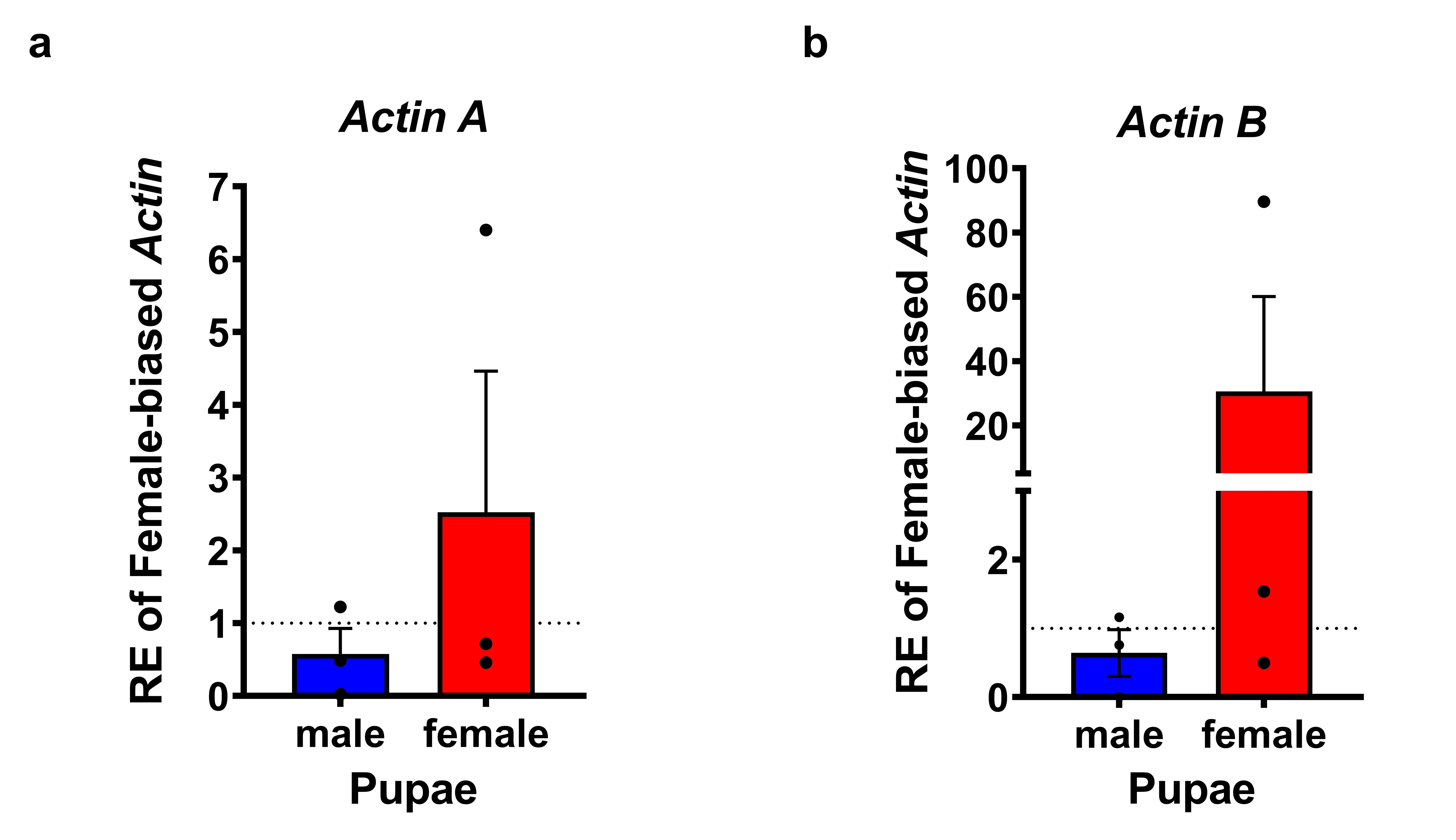
Expression analyses: Real-time expression analysis was done from 3 samples per group, each sample was composed of a pool of 3 male pupae or 3 female pupae. RNA extractions and cDNA synthesis were done as described in previous sections with the RNeasy Mini Kit (Qiagen) and High-Capacity cDNA Reverse transcription kit (Thermo Fischer). Actin housekeeping (HK) (AALB015483) and Ribosomal protein S7 (Rps7) (AALB010399) were used as reference genes and were amplified with the following primers: Actin F (5'-TACAACTCGATCATGAAGTGCGA-3´) and ActinR (5'-CCCGGGTACATGGTGGTACCGCCGGA-3´) and RpS7F (5'AGAACCAGCAGACCACCATC-3') and RpS7R (5'-ACAACCAGCAACGGTTATGT-3´). The q-RT PCR reaction was carried out with the Power Up SYBR green master mix (Thermo Fisher) with 0.33 µM of each primer and 5 µl of cDNA diluted at 1:500 in a final volume of 15 µl. All samples were run in technical triplicate. The genes were amplified as described previously for the tissue-specific qPCR. Delta-delta Ct (∆∆Ct) analysis was performed using the average of Cts of both Rps7 and Actin HK reference genes for analysis of transcript levels of pupae that received actin A dsRNA and Rps7 only for pupae that were fed with Actin B and compared to pupae fed dsRNA of the ant gene as a control. T-test was performed in comparison to the control group.
Bioinformatics analysis
A nucleotide BLAST search using the 500 bp or 600 bp fragment was performed using the VectorBase blast tool with default parameters (66), using the Aedes and Anopheles data sets. Clustal Muscle software was used to align the two actins with gene transcript accession numbers AALB015469-RA and AALB015489-RA (An. albimanus Stecla Gene set AalbS2.6 in VectorBase). The female-biased sequence was aligned with A. darlingi Coari strain (Gene set AdarC3.8 from Vector Base) genomic scaffold_1503 using Clustal muscle software. A homologue search was done for the AALB015469 and AALB014489 genes within the VectorBase gene tab.
Molecular phylogenetic analysis: Phylogenetic tree was built using the Maximum likelihood method based on the Tamura-Nei model (67). Analysis was done with cDNA sequences of the An. albimanus sex-biased actins, An. albimanus Stecla strain actins (Vector base: AALB015481 and AALB015469), Aedes aegypti flight muscle actin-4 (VectorBase: AAEL001951), muscle actin-1 (VectorBase: AAEL001928) and actin-3 (VectorBase: AAEL0094551); Anopheles gambiae actin-4 homolog (VectorBase: AGAP011515) and D. melanogaster flight actin 88F (Ensembl: FBtr0083143). An initial tree was obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. Analyses were conducted using the Mega 7.0.18 software (68).
Data Analysis
One-way ANOVA with Tukey's multiple comparisons, Student's t-tests, and Log-Rank Mantel-Cox test were performed with GraphPad Prism Software version 8.02 (Graphpad Software, CA, USAS). All significant values have a p-value of less than 0.05.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}