All-surviving Ruthenium
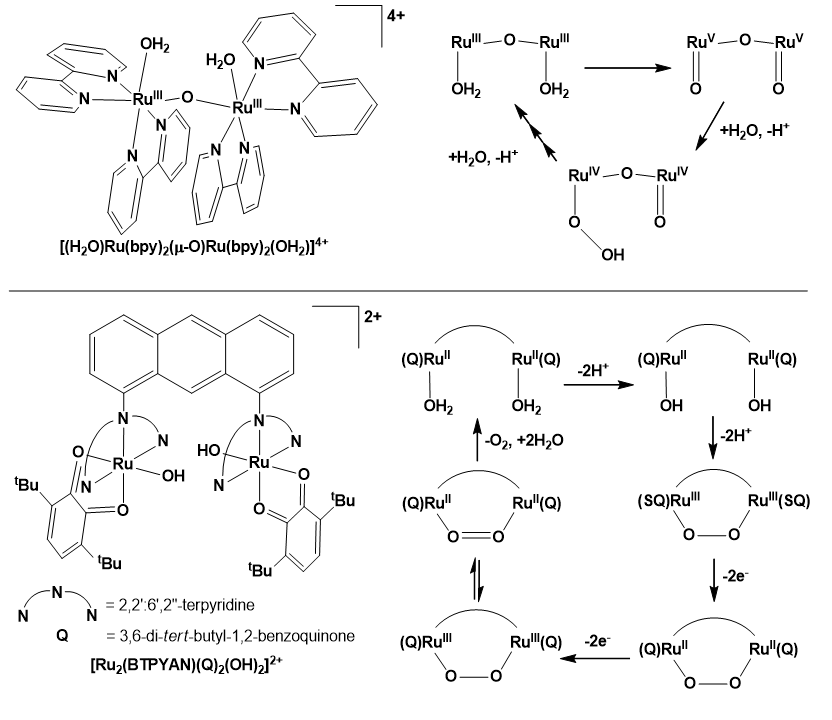
The first thoroughly characterized molecular catalysts for water oxidation were Ru-polypyridyl complexes, starting with the blue dimer cis-[(H2O)RuIII(bpy)2(µ-O)RuIII(bpy)2 (OH2)]4+ (Gersten et al. 1982) (Scheme 6), although an early work also pointed out that oxygen evolution from water is possible using transition metal complexes as catalysts (Elizarova et al. 1981). Ruthenium complexes are robust, inert towards ligand substitution and their higher oxidation states required for water oxidation are indeed accessible, moreover, typically Ru-based catalysts do not require redox active ligands for efficient operation (Matheu et al. 2019).
The water oxidation mechanism by the blue dimer has been subject to a fruitful debate (Liu et al. 2008; Moonshiram et al. 2012) incorporating electrochemical and chemical activation using Ce(IV) in acidic (pH 1), and also photochemical studies at neutral pH using the S2O82−/RuL32+/blue dimer molecular system. In the latter case it has been suggested that the sequential addition of two H2O molecules to the bipyridine ligands may couple to generate O2 (Yamada and Hurst 2000; Cape and Hurst 2008). However, this pathway following the 2e− oxidation of the OH− adducts results in decomposition of bipyridine, ultimately giving CO2 as a product after several redox cycles, since the partly oxidized rings are kinetically susceptible to further oxidation (Liu et al. 2008).
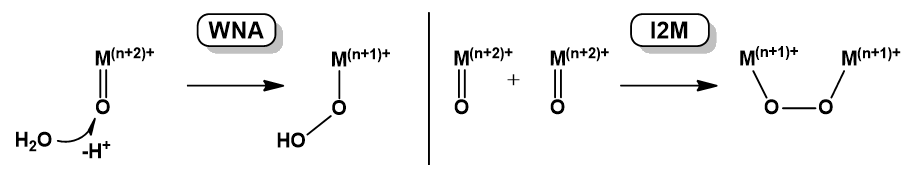
Upon electrochemical activation of Ru catalysts generally the Ru centers and/or the water-derived ligands eject electrons rather than the ancillary ligands. The blue dimer gets oxidized by a 1e− + 3e− sequence. The produced [(O)RuV(bpy)2(µ-O)RuV(bpy)2(O)]4+ complex is thermodynamically competent to form a Ru–O2H intermediate in a WNA step involving one RuV=O unit (Scheme 6). Insight into this mechanism allowed for a strategy for utilizing different [RuII(NN)3]2+ complexes as kinetically facile, external electron-transfer mediators and it was demonstrated that such mediators can enhance the performance of catalytic water oxidation (Concepcion et al. 2008).
Undoubtedly, these achievements inspired several new complexes contributing to the development of mechanistic insights into the molecular catalysis of the OER (Kamdar and Grotjahn 2019). Although ruthenium does not necessitate redox active ligands for catalytic function, quinone ligands were found capable of assisting in Ru-based catalysis (Wada et al. 2001; Kobayashi et al. 2003; Tanaka et al. 2012).
A prominent example is the dinuclear [RuII2(BTPYAN)(Q)2(OH)2]2+ complex (Scheme 6, see abbreviations there) containing two redox active quinone (Q) ligands. The immobilized complex on ITO electrode was capable of electrocatalytic OER at +1.70 V vs. NHE in water (pH 4), producing O2 by 95% charge efficiency and reaching a TON of 6730. Two features were found indispensable to a successful catalysis: the two hydroxide ligands located close to each other due to the structural constraint by the two terpyridine moieties, and the two Q ligands helping intramolecular coupling of the OH ligands to form the O–O bond. Upon the O–O coupling the quinones act as oxidants via intramolecular electron transfer. In this reaction step (SQ)RuII(O-O)RuII(SQ) is formed (SQ is the semiquinonate(-) form of Q) while two protons dissociate from the complex. The SQ ligands undergo oxidation to Q at +0.4 V vs. NHE, furnishing (Q)RuII(O-O)RuII(Q)2+. This is followed by metal oxidation to RuIII at +1.2 V. Valence tautomerism of the produced (Q)RuIII(O-O)RuIII(Q)4+ complex generates (Q)RuII(O=O)RuII(Q)4+ from which O2 liberates upon replacement by two H2O molecules. Note that a theoretical study identified [Ru2(O2−)(Q−1.5)2(BTPYAN)] as a key intermediate and the most reduced catalyst species that is formed by removal of all four protons before 4e− oxidation takes place (Muckerman et al. 2008). The studies on Ru-quinone catalysts signified that the O-O bond formation is attainable at a low oxidation state of the metal and this observation has very important consequences on 1st row TM catalysts.
1st row transition metals
While Ru centers can undergo multiple oxidation steps in a relatively narrow potential range due to effective PCETs, the situation can be very different when water oxidation is carried out using 1st row transition metals. The ligand substitution lability and the access to higher oxidation states demanding high potentials stand as major challenges in the design of catalysts and careful selection of catalytic conditions. Moreover, the release of 4 protons in one catalytic cycle may easily cause ligand protonation and its subsequent dissociation from the metal center as competing process.
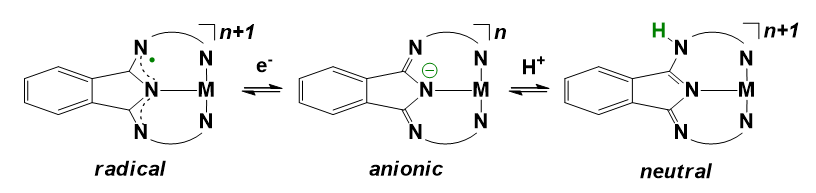
Stabilization of the catalytically active form containing a highly oxidized 1st row TMs center demands strong electron donor ligands. However, this requirement can be circumvented if redox-active ligands are utilized that can be reversibly oxidized to cooperate with the metal center in storing electron vacancies needed for the water oxidation reaction. The involvement of ligand redox activity has been documented in complexes bearing rigid, planar macrocyclic, or semi-macrocyclic ligands with extended π-conjugation capable of carrying an open shell electron configuration. Different families of tetra-amidate ligands were used in Fe (Ellis et al. 2010), Co (Du et al. 2018), Ni (Lee et al. 2020) and Cu (Garrido-Barros et al. 2015, 2017, 2020) complexes.
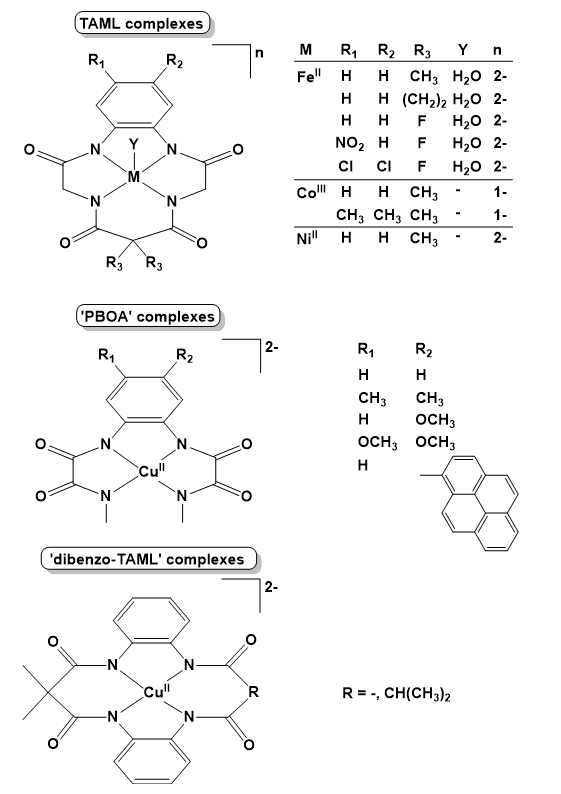
The tetra-amidate macrocyclic ligands in Scheme 7 (TAML complexes) have been studied in details. The first report concerned Fe-TAML complexes that were capable of promoting O2-evolution to a highly variable extent, by mixing them with excess Ce(IV) in water at pH 0.7 (Ellis et al. 2010). Loss of activity was associated with oxidative and hydrolytic inactivation pathways of Fe-TAMLs, also confirmed by control reactions using other Fe-ligand combinations. The rate of the biphasic O2 evolution correlated with the addition of electron-withdrawing substituents to the ligand.
The highest measured TOF was >1.3 s−1 for the TAML with R1 = R2 = Cl and R3 = F, that was sufficiently large that it could be limited by the kinetics of bubble formation and release. Later, formation of a key intermediate, (TAML·)FeV=O with the TAML one-electron oxidized was suggested based on theoretical calculations (Ertem et al. 2012; Liao et al. 2014). In the latter study, this species was proposed to undergo WNA, or nucleophilic attack by a nitrate ion, the water attack being more favored. Notably, according to the results nitrate may function as a co-catalyst by first donating an oxygen atom to the oxo group to form O2 and a nitrite ion, which can then be re-oxidized to regenerate a nitrate ion. Competing pathways were suggested to modify the ligand such as water and nitrate attack as well as ligand amide oxidation, leading to the opening of the benzene ring and fast catalyst degradation.
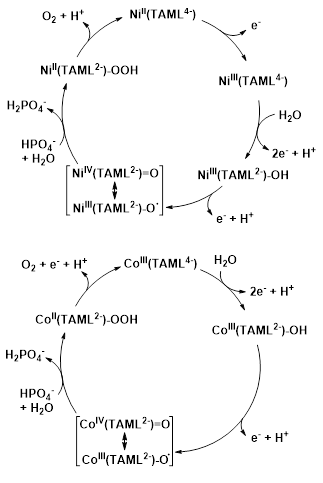
Efficient electrocatalytic OER in neutral aqueous solution was reported by stable CoIII-TAML complexes (Scheme 5) that were confirmed as molecular catalysts under the working conditions (Du et al. 2018). The catalytic cycle was examined in detail by electrochemical methods in combination with DFT calculations. As the working potential was increased, the triplet [CoIII(TAML4−)]− complex was first oxidized to [CoIII(TAML2−)(OH)] via ligand-centered 2e−+H+ PCET in the presence of water. The oxidation of the TAML moiety was clearly indicated by the changes in the C−C bond length of the benzene ring and the N-aryl ring bond lengths, corresponding to the structural change expected in the oxidation of an o-phenylenedicarboxamido ligand to its benzoquinonedicarboxamido form. The resulting [CoIII(TAML2−)(OH)] was further oxidized to [CoIV(TAML2−)=O], which could react with water to form an O−O bond with the help of a buffer anion in a WNA step. The influence of H2PO4− on the WNA process revealed by DFT calculation was in good agreement with the observed buffer effect. Analysis of the calculated structure revealed that both the oxo moiety and the cobalt center bear significant spin densities corresponding to a more correct [CoIII(TAML2−)−O•] assignment. The WNA pathway afforded the quartet hydroperoxo complex, [CoII(TAML2−)−OOH] that was oxidized to the superoxo complex [CoIII(TAML2−)(O2•−)] through a PCET process, to finally release an oxygen molecule and regenerate the resting state [CoIII(TAML4−)]− (Scheme 8). The kcat values calculated for the different complexes were very similar, between 7.53-8.81 s−1, but no catalytic activity could be observed for a complex with a non-redox-active ligand homolog (the benzene ring was substituted by an aliphatic bridge), indicating that the redox-active ligand played a critical role in this multi-electron catalytic cycle. This work underlined the interplay of ligand- and metal-centered redox activity could be a benefit for water oxidation catalysts.
A NiII-TAML (Scheme 7, R3 = CH3) was characterized as electrocatalyst of the OER at neutral pH in phosphate buffer (Lee et al. 2020). The HPO42− anion served as proton acceptor to accelerate the formation of the O–O bond following atom-proton transfer (APT) pathway. The electrochemical activation of the complex started with a reversible, pH-independent NiII to NiIII oxidation at +0.68 V vs. NHE, followed by two irreversible oxidation peaks at +1.03 V and +1.51 V vs. NHE. The first irreversible and pH-dependent oxidation was associated with a 2e−+H+ PCET of [NiIII(TAML4−)] to produce [NiIII(TAML2−)–OH], while the second one was assigned to a [NiIII(TAML2−)–OH] to [NiIV(TAML2−)=O] transition. Oxygen evolution took place at 93% Faraday efficiency and the calculated TOF was 0.32 s−1. The first-order dependence of the catalytic current on catalyst concentration clearly indicated a single-site mechanism. The proposed catalytic cycle is shown in Scheme 8.
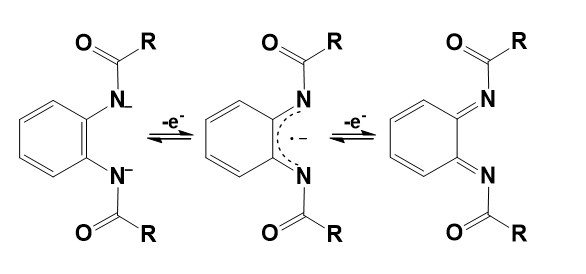
The above catalysts could possibly exploit the one- and two-electron oxidation of the applied TAML ligand according to Scheme 9 thanks to the available formal o-phenylene-/benzosemiquinone-/benzoquinone-dicarboxamido redox forms.
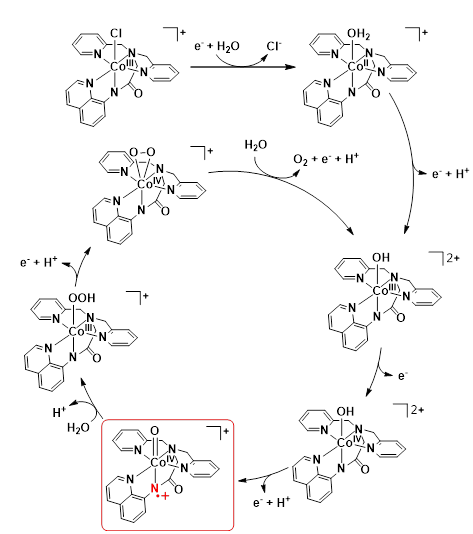
A similar amidate group has been suggested to intervene the redox activation of a mononuclear cobalt(III) complex, [Co(dpaq)(Cl)]Cl featuring the pentadentate ligand, H-dpaq =2-[bis(pyridin-2-ylmethyl)]amino-N-quinolin-8-yl-acetamidate (Biswas et al. 2020). Efficient molecular electrocatalysis was reported to occur at a 500 mV overpotential and pH of 8.0. The redox-active amidate ligand facilitated catalysis through the formation of a reactive oxo-metal species, [CoIV(dpaq•+)(O)]2+ that followed metal oxidation (Scheme 10). According to the mechanism proposal WNA on this putative intermediate formed the O–O bond through a base assisted proton transfer reaction. Interestingly, foot-of-the-wave analysis (FOWA) (Costentin et al. 2012a, b) confirmed the water nucleophilic attack (WNA) mechanism and the high TOFmax value in the order of 104 s−1 obtained by FOWA was comparable to the best performing Ru-based WOCs.
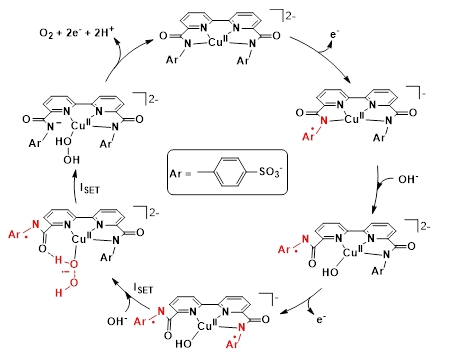
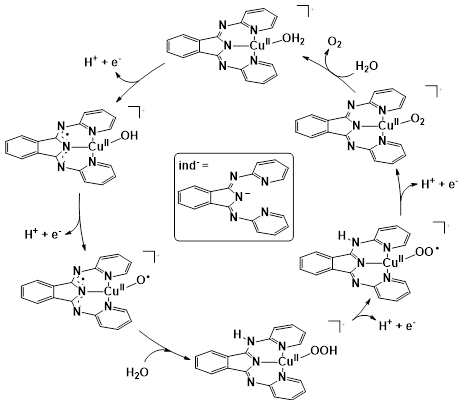
In the case of copper, planar 4N− donor coordination sites like in TAML are well suited for stabilizing an oxidized CuIII metal center and perhaps this metal has been studied most thoroughly with respect to the utilization of redox active ligands. The N1,N1′-(1,2-phenylene)bis(N2-methyloxalamide) (‘PBOA’) ligand family (Scheme 7) demonstrated opportunities in using redox-active ligands to advance Cu-based water oxidation (Garrido-Barros et al. 2015). Introducing electron-donating R1 and R2 substituents at the benzene ring of the PBOA ligand allowed reducing the overpotential in electrocatalytic water oxidation by 530 mV.
The energy of the ligand HOMO correlated with the observed overpotential, indicating that the electron-donating substituents increase the overall energy of the HOMO and favor the oxidation of the ligand. DFT analysis revealed a new mechanism progressing towards the rate determining O−O bond formation in single-electron transformations, and generating a peroxide intermediate with no formal M−O bond (Scheme 11). This has led to a new, general mechanistic proposal, the so-called SET-WNA (single-electron transfer water nucleophilic attack) (Funes-Ardoiz et al. 2017).
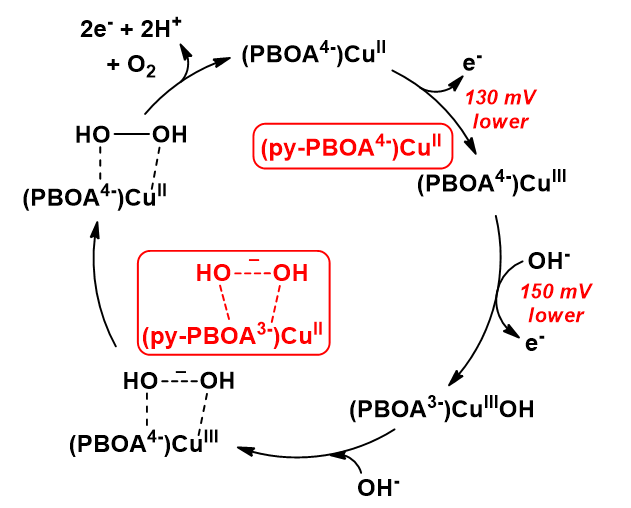
A derived complex utilizing the ligand 4-pyrenyl-PBOA (py-PBOA) was designed to extend the π-conjugation through its structure (Garrido-Barros et al. 2017). The performance of this catalyst (and the parent complex) has been studied in the homogeneous phase and after immobilization by π-stacking to graphene-based electrodes. In the homogeneous phase the electronic perturbation provided by the pyrene substituent reduced the overpotential by 150 mV and increased the catalytic rate by more than 20 times to achieve a kcat of 128 s−1. Based on spectroscopic investigations on the one-electron oxidized form, the authors addressed the oxidation process to the pyrene moiety in aqueous solution in contrast to the parent complex, in which the oxidation remained metal-centered (the differences are highlighted in red in Scheme 11). This difference was associated with the lower oxidation potential at the ligand site due to the presence of the pyrene group together with the large stabilization to the putatively charged oxidized species in the aqueous environment. Immobilization on a graphene surface provided additional delocalization that improved the catalytic performance of both catalysts, however, the py-PBOA complex was more active. The overpotential of 538 mV, kobs of 540 s−1 and TON > 5300 that by a rational design stability of first row TM catalysts can be dramatically increased.
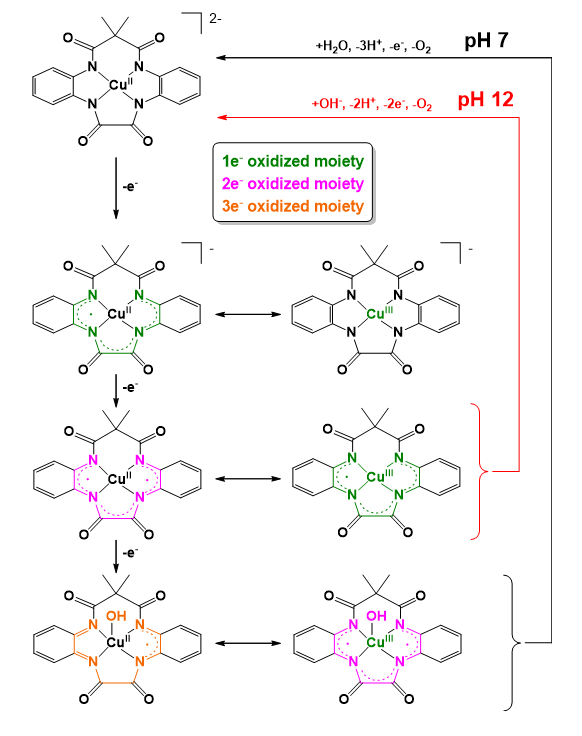
The same group underlined in a later work how crucial it is to elucidate the actual sites affected by consecutive oxidations yielding the different formal oxidation states of the complex and how subtle changes can perturb the delocalization of electron density. For this purpose they developed two new ligands with 13- and 14-membered rings that can be regarded as ’dibenzo-tetraamido macrocycles’ (’dibenzo-TAML’ in Scheme 7) (Garrido-Barros et al. 2020) in addition to the above discussed semi-macrocyclic ‘PBOA’ ones. They found that a mere one-atom change in the ring size and change in solvent polarity were enough to shift the delocalization of electron density from the metal over the ligand π-system. Valence tautomerism exerted by the solvent interaction ‘evidenced the energetic near degeneration in the frontier orbitals and the subsequent easy access to the different oxidation states, corresponding to metal-centered, d-orbital oxidation or ligand centered oxidation’. For example, the coordinated 13-membered ‘dibenzo-TAML’ was involved in two 1e− oxidations due to the high delocalization of spins (Scheme 12) resulting in two energetically levelled oxidation steps within an 80 mV potential window. In contrast, this metal-ligand redox cooperativity was missing from the complex with the 14-membered ‘dibenzo-TAML’ macrocycle (Scheme 5, R = CH(CH3)2), where the π-conjugation between both phenylene moieties was broken due to the presence of two dimethylmalonamide bridges with sp3 carbons and the saddle distortion of the ligand.
These subtle changes in the ligand structure fundamentally determined the fate of the catalysts upon electrolytic water oxidation, since the stability of the different spin states of the complex at the different oxidation states relied strongly on the extent of π-delocalization. The complex bearing the larger macrocycle ligand decomposed during catalysis, whereas the one containing the 13-membered macrocycle (Scheme 12) remained stable and highly active thanks to the evidenced redox cooperativity between the metal and the ligand. The highly oxidized reactive form thus has been stabilized due to the delocalization of the accumulated oxidative equivalents (the proposed mechanism and charge delocalization in the different oxidation states are shown in Scheme 12). Importantly, this way the high-energy oxidized states centered only at the ligand, or the metal center have been avoided so that both ligand oxidative degradation and high-valent metal centers requiring large potentials and frequently involved in formation of metal oxides were absent. In addition, the two ligand-based oxidations in a narrow potential window were concluded to be essential to enable water oxidation at neutral pH, where an extra oxidation is required to generate a more active Cu complex. As a result, at neutral pH catalysis proceeded with a kobs of 140 s−1 at an overpotential of only 200 mV.
A comparison was also made in this context with an earlier catalyst featuring electronic delocalization limited to single phenylene moiety (Scheme 7, PBOA ligand, R1 = R2 = H) (Garrido-Barros et al. 2015). In this case, water oxidation at pH 12 proceeded via metal oxidation followed by ligand oxidation as with the macrocycle (Scheme 12). However, the acyclic ligand-containing complex was prone to acidic demetallation below pH 10 that prevented its application in water oxidation at neutral pH, in sharp contrast to the macrocyclic complex. This observation underlined the importance of the cooperative metal-ligand design approach for oxidative equivalent accumulation to find more stable and efficient molecular catalyst for redox reactions. The authors also emphasized that attention should be paid to the analysis of the irreversible pre-catalytic electrochemical oxidation features that may hinder substantial changes in the pre-catalysts upon oxidation and may indicate degradation.
Following simple logic we can conclude that shutting down the redox activity of the metal center in parallel with triggering that of the ligand is possible by appropriate ligand design. Indeed, recently a new family of dianionic [2,2’-bipyridine]-6,6’-dicarboxamide ligands (Scheme 13), substituted with redox-active phenyl or naphthyl moieties have been described (Gil-Sepulcre et al. 2021). The electrocatalytic OER process involving only ligand-based redox events was a consequence of two features of the ligand. First, a CuIII/II redox couple at relatively low potentials necessitates highly anionic, stronglyσ- and π-donating ligand environment coordinated to the Cu center. In the case the bpy-dicarboxamide ligands two neutral N donors of the bpy and two anionic N donors of the amide groups had insufficient electron-donating capacity to stabilize the oxidized metal center below or at the onset potential of catalysis. The second feature was that two redox-responsive phenyl/naphthyl groups were attached to the ligand conjugated with the amide donors that were able to accommodate two distal electron vacancies. Upon oxidation, one of the aryl-amide groups could be detached from the metal, opening a quasi-equatorial coordination site for an OH− ligand.
A detailed electrochemical analysis at pH 11.6 revealed a large electrocatalytic current associated with O2-evolution at an overpotential of 830 mV for the complex shown in Scheme 13. Experimental and computational results supported a catalytic cycle progressing towards the active form through single-electron transfer steps corresponding to a SET-WNA mechanism, including intramolecular single-electron transfer (ISET) steps (Scheme 13). Upon the first oxidation a triplet CuII complex is formed with a radical cation mainly centered on one of the aryl-amidate moieties. Evidence for this possibly rate limiting ligand-based electron transfer in electrocatalytic OER was provided by the correlation between the substituent effect on the aryl groups and the observed onset potential of catalysis. Accordingly, the kobs values were in the range of 5–35 s−1 and the Faraday efficiencies were between 40 and 76%, both related to the structure of the ligand and the stability of the oxidized moiety. The Cu center served as a scaffold for the two bpy-amide groups bonded to the metal center, and at the same time OH− coordination and activation could take place. According to the proposed mechanism this Cu-OH group can be attacked by an external hydroxide via a radical-nucleophilic pathway, generating a Cu-(HOOH) species. Since this process involves ISET event between the aryl radical cation and the Cu-OH group, ‘the metal center is responsible for placing the two groups sufficiently close so that fast ISET can occur’ (Gil-Sepulcre et al. 2021). The role for the aryl substituent was clearly discussed both from a thermodynamic perspective, because it tuned the overpotential for OER catalysis, and from a kinetic perspective, ‘because of the low reorganization energy of extended polyacenes’ upon electron transfer ensuring fast kinetic processes. Considering the high versatility in ligand design, the low efficiency obtained for the above complexes that is due to competitive deactivation pathways can be solved in the future.
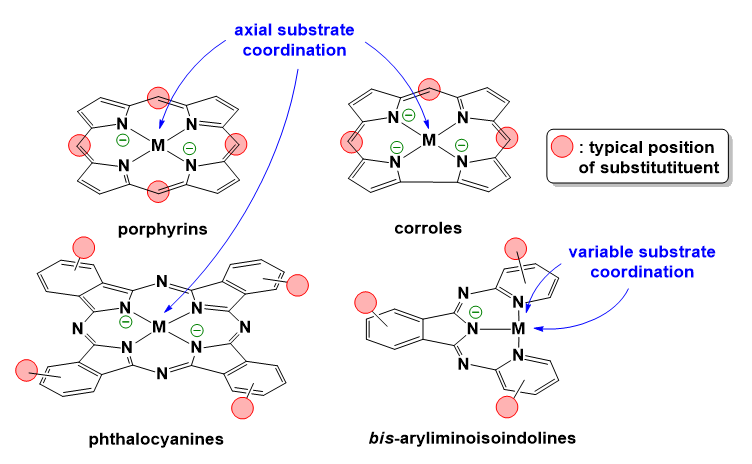
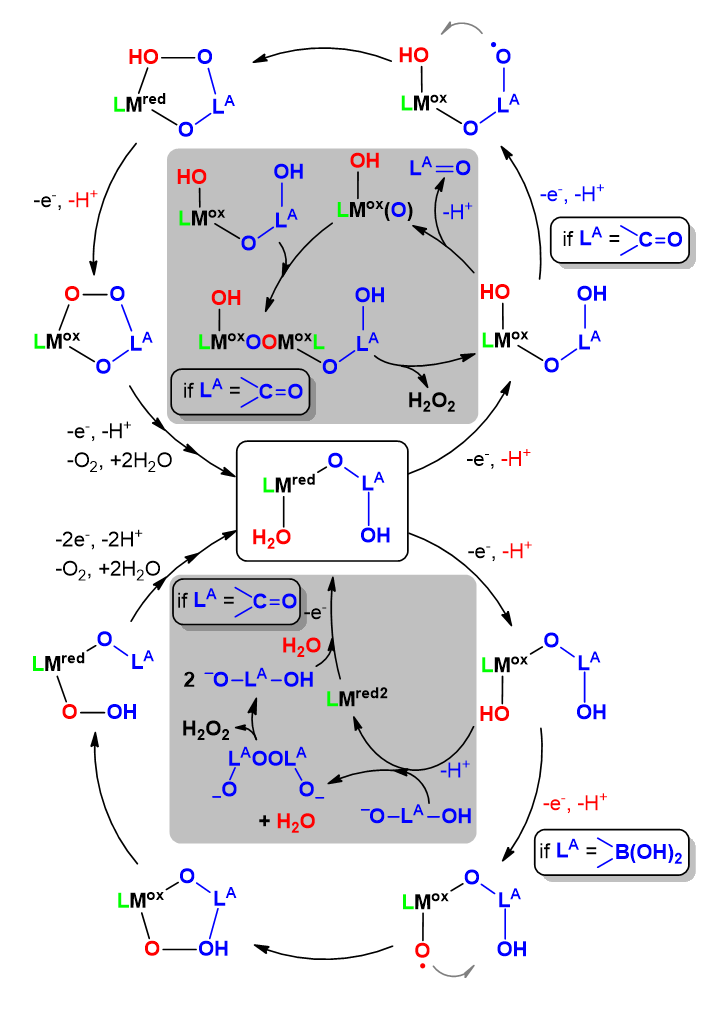
Due to their cyclic π-delocalization porphyrins and corroles are capable of stabilizing electron vacancies. In addition, the two or three negative charges at the N donor groups, respectively, can stabilize the coordinated metal center in higher oxidation states. Thus, these compounds represent an exciting redox non-innocent and potentially redox-active ligand platform with rich possibilities in derivatization as indicated in Scheme 14. Their advantages have been discussed earlier (Zhang et al. 2017b). In context with the present overview, we emphasize here that metalloporphyrin catalysts for the OER are generally proposed to undergo first a 1e− oxidation concerning the ligand that is followed by 1e− oxidation of the metal center.
Metallocorroles can behave in a similar fashion upon oxidative activation of the OER catalysts (Zhang et al. 2017b). In metallocorroles the stabilization of high-valent metal ions by corroles is viable through a combination of short metal-nitrogen bonds and large metal out-of-plane displacements (Gross 2001). A key difference from metalloporphyrins is the wider prevalence of non-innocent electronic structures and full-fledged corrole•2− radicals among metallocorroles (Ghosh 2017). There is gathering evidence that the non-innocent or redox-active behavior of this ligand family is prevalent (Lemon et al. 2016; Schöfberger et al. 2016; Sinha et al. 2018, 2020; Garai et al. 2018; Mondal et al. 2020).
Importantly, these ligand architectures are suitable for surface immobilization through covalent linkers or axial coordination of the metal center by tether ligands grafted onto the surface, or by exploiting their high adsorptivity (Schöfberger et al. 2016), that makes their utilization in (photo)electrodes directly available (Zhang et al. 2017b). Two-electron oxidation of water to form hydrogen peroxide by using metalloporphyrins containing redox silent metal centers like Al, or Sn has been also addressed as viable alternative strategy for artificial photosynthesis systems (Kuttassery et al. 2018) as well as light-driven oxidation of halide anions by metallocorroles (Mahammed and Gross 2015).
Phthalocyanines (PCs) are expected to exhibit advantages like those of porphyrins and corroles in OER catalysis (Scheme 14). For example, photocatalytic water oxidation in the presence of a water soluble CuII-PC complex was proposed to follow an I2M mechanism in borate buffer at pH 9.5 (Terao et al. 2016). According to computations, upon 1e− oxidation of the complex a PC radical is generated instead of a metal-based electron loss, thus the CuIII oxidation state generally leading to complex photodegradation does not occur. Importantly, competitive ligand exchange between chloride ions and water molecules for the fifth coordination site inhibited the reaction, calling attention to the potential detrimental effect of impurities.
Recent findings on the possible application of metallo-PCs are encouraging. Cu-PC was reported to act as efficient redox mediator for TiO2 nanorod-based PEC water splitting (Li et al. 2019), moreover, conjugated to carbon quantum dots (CQDs) by π–stacking and then coupled with BiVO4 to construct a hybrid water oxidation photocatalyst BiVO4/CQDs/CuPC (Xu et al. 2021). A somewhat similar system with Co-PC deployed on BiVO4 was also very recently reported to enhance the photocurrent in PEC oxygen evolution (Shen et al. 2021). Polymeric copper and cobalt PCs (Cu-pPC and Co-pPC) were prepared as thin films on FTO from evaporated metal films via a chemical vapor deposition (CVD) process with 1,2,4,5-tetracyanobenzene and successfully operated in 0.1 M KOH electrolyte to generate O2 electrocatalytically (Geis et al. 2016). These and other types of polymers, like metallated azo-naphthalene diimide based redox-active porous organic polymer frameworks (Bhat et al. 2018) are encouraging to envision future applications of such materials. Note on the other hand that the example of a MnII-PC complex resulting in MnOx formation upon electrocatalytic water oxidation in unbuffered acetonitrile-water mixture shows that organic ligand platforms may not be suitable under harsh conditions and may undergo hydrolytic degradation (Mousazade et al. 2019).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}