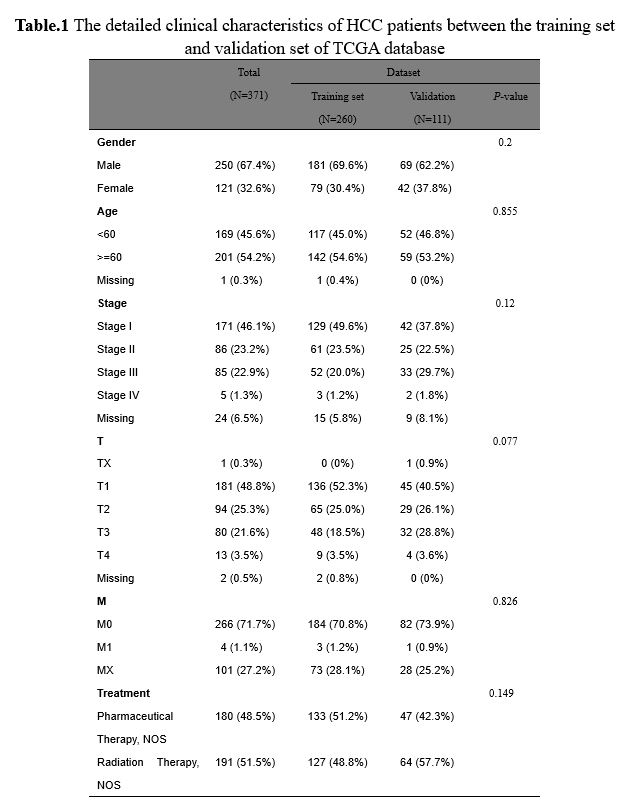
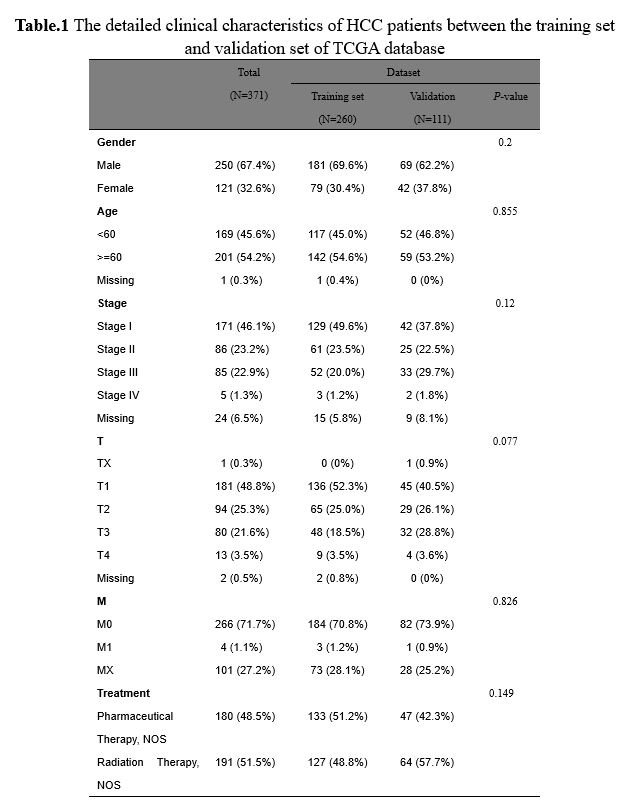
Identification of co-differentially expressed GT genes in HCC
A PCA of 421 samples in the TCGA database was performed, and the results revealed that there was an obvious difference between tumor tissues and normal tissues samples, which was applied for the subsequent analyses (Fig.2A). To further explore which GT genes may act as prognostic factors in the progression of HCC, we first comprehensively analyzed the training set in the TCGA database of HCC. As presented in Fig.2B, a total of 2264 DEGs were screened based on the threshold of |log2FC| ≥ 1 and P-value ≤ 0.05, of which 1765 were up-regulated and 499 were down-regulated compared with the tumor group (Supplementary Table.3). The heatmap showed the expressions of DEGs between tumor group and normal group (Fig.2C). Importantly, the expressions of the top10 DEGs were statistically significant differences between the two groups (Fig.2D, P < 0.05). Furthermore, these DEGs were combined with 210 GTs downloaded from literature, and 28 co-differentially expressed GT genes were obtained in the TCGA database of HCC (Fig.2E).
Identification of gene signatures associated with OS in HCC
To further identify the gene signatures in HCC prognosis, we undertook a univariate Cox regression analysis on the 28 co-differentially expressed GT genes. As a result, 11 of them were obtained with the cut-off of P-value < 0.05 (Fig.3A). After the least absolute shrinkage and selection operator (LASSO) Cox regression algorithm in the TCGA dataset, we obtained 4 gene signatures, such as ALG3, B3GAT3, GLA, ST6GALNAC4 (Fig.3B-C). To better demonstrate the prognostic role of these 4 gene signatures in HCC, we firstly used K-M analysis in the TCGA dataset in which samples were divided into high and low expression based on the gene expression level (Supplementary Figure.1). All 4 of these genes were significantly associated with the OS of HCC patients, which indicated that these gene signatures may be acted as prognostic factors in HCC. Intriguingly, PPI network revealed that there was no direct interactions between 4 gene signatures (Fig.3D).
Prognostic value of gene signatures in the training dataset of TCGA database
Having shown that these gene signatures may act as prognostic factors in the progression of HCC, we attempted to investigate their prognostic value in HCC. Therefore, we constructed the prognostic model of gene signatures in the training dataset of TCGA database. The risk score was calculated according to the gene expression levels and their corresponding coefficients. Based on the median value of risk score, the patients with HCC were divided into high- and low-risk groups (Fig.4A). The results showed that the number of patients died of HCC was significantly increased with the increasing risk score. The OS of HCC patients had an obvious difference between the two risk groups (Fig.4B, P=0.00095). Thereafter, ROC curve was performed to investigate the effectiveness of the prognostic model (Fig.4C). The area under the curve (AUC) reached 0.676 at 3 years, 0.631 at 5 years, and 0.539 at 7 years, which indicated that the risk score of prognostic model had high accuracy. A nomogram of 3-year and 5-year survival probability was displayed in Fig.4D. In addition, the calibration plots of this risk prognostic model were further constructed (Fig.4E-F, c-index=0.66328, calibrated c-index=0.64057)
Validation of gene signatures in the validation set of TCGA database
To further seek additional evidence that the gene signatures are prognostic factors, we generated a prognostic model in the validation dataset of TCGA database. Interestingly, survival analyses of 4 gene signatures confirmed that two of these genes were correlated with poor OS of HCC (Supplementary Figure.2). To demonstrate the robustness of the validation model from the TCGA database, the patients with HCC were divided into high- and low-risk groups according to the median value of risk score (Fig.5A). Consistent with the results obtained from the training dataset of TCGA database (Fig.5B, P=0.0039). Besides, the ROC curve showed that the AUC of 4 gene signatures reached 0.708 at 3 years, 0.712 at 5 years, and 0.594 at 7 years, further revealing the accuracy of risk score in the validation prognostic model (Fig.5C).
The correlation of the risk scores and clinicopathological characteristics in patients with HCC
The expressions of the 4 gene signatures between the two risk groups and clinical characteristics in the TCGA database were visualized by heatmap (Fig.6A-B). We found that the expressions of these gene signatures were significantly increased as the increasing risk score in the TCGA database. Furthermore, we performed univariate and multivariate Cox regression analyses after adding clinical characteristics to investigate whether the risk score of prognostic model was an independent factor for the HCC prognosis (Fig.6C-D). In the univariate Cox regression analyses, the risk score was correlated with the OS of HCC both in the training set and validation set of TCGA database (TCGA training set: HR= 2, 95% CI =1.5-2.8, P=2.2e-05; TCGA validation set: HR= 2, 95% CI =1.4-2.8, P= 0.0034), respectively. The multivariate Cox regression analysis showed that the risk score of prognostic model was still proved to be an independent factor for the OS of HCC patients (TCGA training set: HR =2.2, 95% CI = 1.5-3.2, P=5.4e-05; TCGA validation set: HR=1.7, 95% CI = 1.1-2.7, P = 0.018).
Functional analyses in the TCGA database of HCC
Based on these observations, we next attempted to elucidate the biological progresses related to the risk score. Therefore, we focused on the DEGs between the two risk groups to perform GSEA. As a result, GO analysis of the DEGs were involved in several immune-related biological processes, such as GO:0002263 cell activation involved in immune response, GO: 0002275 myeloid cell activation involved in immune response, GO:0002283 neutrophil activation involved in immune response and GO:0002520 immune system development, which may be highly associated with the development of HCC (Fig.7A, P-value < 0.05). All significantly enriched GO terms were shown in Supplementary Table.4.
In order to investigate whether the risk score was correlated with the immune status in HCC, we analyzed the differences between the high-risk and low-risk groups by quantifying the scores of immune cells enrichment. Interestingly, activated CD4 T cell, activated dendritic cell, CD56dim natural killer cell, central memory CD8 T cell, Eosinophil, MDSC, Natural killer T cell showed obvious significantly differences between the two risk groups (Fig.7B, *P < 0.05, **P < 0.01,***P < 0.001, ****, P< 0.0001). Moreover, the immune score calculated by ESTIMATE algorithm in high risk group was higher than those of low risk group, which further revealed that the risk score of prognostic model was strongly correlated with the immune status of HCC patients (Fig.7C, **P < 0.01). Interestingly, the stromal score and ESTIMATE score showed no significant difference between high-risk and low-risk groups (Fig.7D-E).
{kind=link}
{kind=link}