Thermal evolution of the crystal structures: bond distances and dihedral angles.
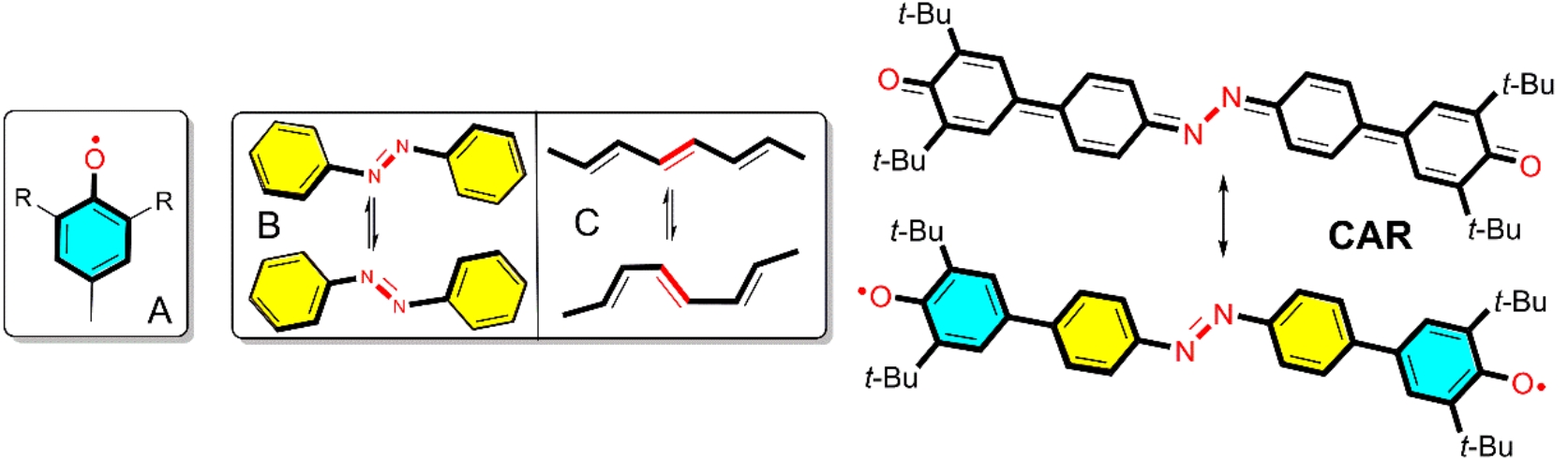
The detail of synthesis and characterization of CARH (non-diradical fully aromatic CAR analogue) and CAR were described (Supplementary Figs. 1–3). Single crystals of CARH and CAR were grown by using the slow solvent evaporation method in dichloromethane solution under N2 atmosphere. The structure of CAR was studied at several temperatures with main focus in those at 130, 290 and 340 K. The average R factor of these structures is around 6.5%, which is reliable for the forthcoming discussion. The selected bond lengths for CARH and CAR are shown in Figs. 1 and 2 (Supplementary Fig. 4 and Tables 1–6).
The following structural features are highlighted. At 130 K, in Fig. 1: i) the two phenoxyl substituents in CARH are twisted regarding the central azobenzene core with a dihedral angle of 38.76° whereas this angle is 5.78° in CAR; ii) the carbon-oxygen bond length in CAR (1.245 Å) is shorter than that in CARH (1.377 Å), which is very close to a typical carbon-oxygen double bond of p-terphenoquinone (1.231 Å)15; iii) a significant bond length alternation of r2, r3, r4 in the two phenoxyl benzenoid rings is observed; and iv) a crucial change of the nitrogen-nitrogen bond r10 length from 1.252 Å in CARH to 1.300 Å in CAR, indicating its partial nitrogen-nitrogen single bond character. All these features indicate a sort of quinoidal-like (hereafter, Q) structure on the phenoxyls of CAR at 130 K.
At 290 K: i) the single crystal structure reveals that the nitrogen-nitrogen bond r10 (1.257 Å) becomes much closer to that of CARH (1.252 Å); ii) the bond length alternations of r6, r7, r8 in the internal benzene rings become smaller; iii) the carbon-carbon bond r5 (1.478 Å) in CAR is almost identical to that of CARH (1.484 Å); and iv) the carbon-oxygen r1 and carbon-carbon bonds r2, r3, r4 do not show significant changes at 290 K. These all suggest an exclusive transformation of the azobenzene core at 290 K attaining a structure closer to that typical of aromatic azobenzene. Overall, the bond length changes from 130 to 290 K are mainly in the azobenzene core documenting a decreasing quinoidal character in favor of a more aromatic-like shape (hereafter, A). This type of transformation is typically found in diradicals and can be termed as a “normal” quinoidal→aromatic step .
Surprisingly, the most significant changes at 340 K compared to 290 K are: i) the length of r5 is 0.029 Å shorter than at 290 K (similar to that at 130 K); and ii) the r7 CC bond length decreases by 0.024 Å becoming similar again to that at 130 K. This 290→340 K changes indicate the regaining of partial quinoid character at 340 K or pseudo-quinoidal structure (hereafter, PQ). In diradicals and in π-conjugated molecules, a continuum in the “normal” quinoidal→aromatic transformation is always observed on heating, what makes certainly unexpected this structural A→PQ inversion, a new and, to the best of our knowledge, unknown “reversed” quinoidal→aromatic transformation we discover in CAR.
The thermal variation of the dihedral angles in Fig. 2 (Supplementary Fig. 5) uncovers two distinctive effects. First, an inter-ring torsional motion as the temperature increases to 290 K in which dihedral angles between the connected phenoxyl and azobenzene rings vary: for instance, from 5.78º at 130 K to 8.96º at 290 K, and then a reduction up to 8.36º passing to 340 K. Second, the two terminal phenoxyls are twisted in opposite ways with respect to the central azobenzene thereby assuming the same absolute angle value but opposite signs. The simultaneous presence of the same terminal group twisted in opposite directions in the same molecule and crystal environment, at the same time associated with the opposite orientation of the N = N group suggests the active role of the bicycle pedal motion well known in crystals of azobenzene derivatives.13 This dihedral angle evolution delineates a combined effect of the pedal motion jointly acting with the phenoxyl twist owing to the existence of rotational mobility of the azobenzene core which is expected to increase with temperature. In line with this, the crystal structure also shows that the CCNN dihedral α, associated with the bicycle pedal motion of the two CC adjacent to the central NN, changes sign with temperature: although the value is always small, the sign change further corroborates the rotational mobility.
Physical properties. Magnetic susceptibility of solid CAR was measured using a Quantum Design MPMS 5XL SQUID magnetometer and a value of -0.000295 emu mol− 1 was used as Pascal’s correction16 for diamagnetism of the sample and holder (Supplementary Fig. 6). The plot of χT-T in Fig. 3a shows a curvilinear relationship, Furthermore, the χT value (0.35 emu K mol− 1) of CAR at 300 K is smaller than the theoretical value of an ideal diradical (0.75 emu K mol− 1) indicating antiferromagnetic coupling in the ground electronic state. By fitting the curves with a modified Bleaney-Bowers Eq. 17, the single-triplet energy gap (ΔEST) amounted to −1.31 kcal mol− 1
The ESR spectra (Fig. 3b) showed characteristic triplet patterns at 130 K arising from thermally populated triplets. These are: i) ESR resonances are split by the zero-field splitting; and ii) the unique half-field forbidden transition triplet signal (Δms = ± 2) at 168 mT was observed at 130 K (Fig. 3a inset) which is rarely found in thermally excited triplets of organic singlet diradicals.18 The zero-field splitting parameters amount to D = 0.00503 cm− 1 and E = 0.00013 cm− 1 from the spectrum simulation. From the D value, the average distance between the two triplet unpaired electrons (D ~ 1/d3) was calculated as d = 8.04 Å, which is shorter than the length of azobenzene moiety (9.094 Å) suggesting that the spin density is mostly in the molecular center at 130 K in agreement with the remaining phenyls attaining a more quinoidal shape (Q form). Variable-temperature ESR measurements were also performed (Supplementary Fig. 7) and the ΔEST was estimated to be −3.41 kcal mol− 1 by fitting the curves with the Bleaney-Bowers equation (Supplementary Fig. 8), which agrees with the SQUID result.
The solid-state optical absorption spectrum of CAR in Fig. 3c (Supplementary Fig. 9) shows two weak bands at 888 and 997 nm due to the S0→S1 transition to a dark double exciton state typically arising in diradicals. 19 The strongest band owing to the S0→S2 excitation is at 645 nm. Figure 3d represents the Raman spectra of CAR taken with the Raman excitation laser lines at 633 nm (in resonance with the 645 nm absorption) and at 1064 nm (in pre-resonance with the 997 nm band). The two spectra share the bands at 1597 and 1563 cm− 1 (blue shaded 1 and 2 bands in Fig. 3d) while the 1064 nm spectrum have additional bands at 1540 − 1530 cm− 1 (red 3 bands in Fig. 3d) which vibrationally identifies the double exciton state of the CAR diradical.
Singlet-triplet gaps ( ΔE ST ) and thermal spin distribution. To further understand the magnetic structure of CAR, DFT calculations were carried out at B3LYP/6-31G(d) and M06-2X/6-311G(d) levels (Suplementary Tables S7 and S8).20–24 Considering CAR in isolated in the vacuum, an open-shell singlet ground state and a ΔEST gaps of −0.43 kcal mol− 1 (M06-2X/6-311G(d) and −0.94 kcal mol− 1 (UB3LYP/6-31G(d)) are predicted. On the x-ray molecular structures at 130, 290 and 340 K, UB3LYP/6-31G(d) quantum chemical calculations predict, respectively, three different ΔEST values of −2.62, −0.80 and −1.20 kcal mol− 1 which, on average, yields a value of −1.54 kcal mol− 1 very close to the −1.31 kcal mol− 1 from SQUID measurements. Furthermore, a diradical character value of y0 = 0.93 is estimated for the in vacuum broken-symmetry (BS) optimized structure (M06-2X/6-311G(d) level); whereas values of y0 = 0.81, y0 = 0.92 and y0 = 0.88 were calculated for the crystal structures of CAR at 130, 290 and 340 K, respectively. This theoretical description indicates the modulation and tuning of the diradical character of CAR in its thermal forms referred as Q form (y0 = 0.81), A form (y0 = 0.92) and PQ form (y0 = 0.88). Hence, CAR has three singlet-triplet excitation processes due to the progressive thermal conversion of its structure. Unfortunately, these three ΔEST gaps could not be resolved experimentally by SQUID magnetometry which overall detects one single process.
In the crystal structures at different temperatures, spin distributions were calculated (M06-2X/6-311G(d)) and selected spin population values of specific atoms shown in Fig. 4a (Supplementary Fig. 10 and Table 9). As the temperature increases from 130 to 290 K, the spin density decreases in the azobenzene core (Fig. 4b) and moderately increases in the phenoxyls. The most obvious change of spin density lies in the N atom which decreases from 0.174 to 0.081 from 130 to 290 K and concomitantly the spin density of the azobenzene core become very small. At 340 K, the integrated spin density of the N atoms and of the azobenzene unexpectedly increase. Correlating spin distribution and molecular structures, at 130 K the Q form spreads out the spin density more or less uniformly over the whole skeleton of CAR (light blue circles and red lines in Fig. 4). The transformation into the A form at 290 K largely concentrates the spin density in the terminal phenoxyls (red circles and black lines in Fig. 4) while at 340 K, the PQ structure again redistributes the spin density towards the central moiety.
Mechanism of spin delocalization.
The paths of transformation among different spin-delocalized structures. The thermal transformation of CAR along the y0 coordinate corresponds to two separated processes, an increase (0.81→0.92) for Q→A followed by a decrease (0.92→0.88) for A→PQ wherein the latter step cannot be viewed as a way back of the former. This is deduced from calculations of the Huang-Rhys factors25 alongside the two above structural transitions (Supplementary Table 10) which reveal that the set of vibrational normal mode contributions that conduct the two transformations are different (e.g., there is not a vibrational reaction coordinate that sequentially transforms among them). Hence, the portion of the potential energy surface (PES) driving the structural changes of CAR may be figured out in a multidimensional space in which not only stretching coordinates but critically here torsional nuclear displacements, more likely affected by the solid-state environment, are involved. The multidimensional character of reaction coordinates of CAR agrees with the simultaneous changes observed experimentally for the π-bond distances and for the dihedral angles from x-ray data.
The effect of the crystal in the spin transformation: QM/MM calculations. Given the distinctive role of torsional motion, the PES of CAR was explored with QM/MM calculations (Supplementary Figs. 11, 12, 13 and 14) as a function of: i) the inter-ring torsion around the CC bonds connecting the azobenzene moiety with the phenoxyl terminal units (Fig. 5a), and ii) the bicycle pedal motion involving the two CC bonds adjacent to the central azo group (Fig. 5b). The three crystal geometries at 130, 290 and 340 K were considered inside a cluster formed by 18 molecules that completely surround the central one, described at QM level, to include the effects (via MM potential) of the nearby molecules in the crystal.
The simultaneous torsion of the two CC bonds may occur by twisting the whole azobenzene moiety inside the crystal cavity with respect to the terminal phenoxyls which remain almost positionally cleaved (dumbbell structure), such as drawn schematically in Fig. 5. Similarly, also the bicycle pedal motion in Fig. 5 can occur by leaving the terminal moieties of the CAR molecule almost fixed. We thus realize that these two motions are unique to jointly provide with torsional solid-state mobility to azobenzene.
A key point is that the QM/MM computed PES along the two Q→A and A→PQ selected torsional paths are asymmetric (Fig. 6 and Supplementary Fig. 12–14) with respect to the planar structure in contrast with the isolated/in vacuum computed PES which is symmetric with two degenerate energy minima separated by a small barrier (i.e., such as in biphenyl)26. The asymmetry is more marked for the torsion of the whole azobenzene with respect to the phenoxyl groups (Fig. 6) compared to the bicycle pedal motion as determined by the interaction with neighbor molecules in the crystal, which are kept fixed while scanning the PES. The PESs alongside the 130 to 290 K transformation do not intercept each other by varying the torsional angle (Supplementary Fig. 12) indicating that the Q→A transformation is progressive and might be driven by entropy effects based on the increase of molar entropy on going towards a more distorted structure. The increase of entropy is conducted by thermal excitation/population of low energy torsional modes which is not reflected in the PESs behavior.
The consideration of the 290 and 340 K PES´s shows a crossing around 30 º in which the A form is favored for θ > 30 º and the PQ form is preferred for θ < 30 º. This crossing could be favored and modulated by coupling with the bicycle pedal motion. In fact, if we look at a CAR molecule inside the crystal, and focus on the two closest CAR molecules (above and below the central one in Supplementary Fig. 5), the θ angle is found positive or negative only when coupled with a specific orientation of the azo group. This suggests that the torsion of the azobenzene may be coupled with the bicycle pedal motion. Therefore, it is the joint action of the torsion of azobenzene and of the bicycle pedal motion that assists the crossing between PESs and drives the geometry change occurring with the temperature increase, such as schematized in Fig. 5. The A→PQ transition takes place as a way to reduce the interaction energy in a given interval of dihedrals.
Taking into account the distinctive out-of-plane topologies of the twisting and bicycle pedal motions, the observed transformations can be rationalized: i) the “normal” Q→A change is exclusively driven by low energy torsions which always favors the aromatic structures; however, ii) when the higher energy bicycle pedal vibrational states are excited together with the torsional vibrations, a less distorted thermalized molecular structure, PQ, with smaller diradical character is preferred, outlining a “reversed” quinoidal→aromatic transformation.
{kind=link}