By a conservative estimate, there are at least a billion species of bacteria, but only 30,000 are formally named.(21) Of those, less than 2% can be cultured and identified in the laboratory.(22) However in the 1980’s, the introduction of the polymerase chain reaction (PCR) targeting the highly conserved ribosomal genes (16S rRNA) of bacteria allowed for identification of unculturable organisms.(23) Subsequent advances in DNA sequencing and other molecular techniques have allowed for inexpensive, rapid culture-independent identification of the vast array of resident microbiota in health (normobiosis or eubiosis), and in disease (dysbiosis), such as cancer. Despite the explosion in microbiome studies primarily in the gut looking for culprit bacteria that may cause malignancies, especially colon cancer and other gastrointestinal diseases, results have been disappointing since findings are inconsistent and not reproducible, likely due to variability of gut microbiota depending upon the sex, race, age, geographic location and lifestyle factors of diet, exposures, drugs and exercise.(24)
Although in preclinical models, the bacterial composition of the gut microbiome appears to determine whether there is a response to immune checkpoint inhibitors (ICI), numerous human studies have failed to identify specific species or phyla that are clearly associated with immunotherapy efficacy in any cancer.(24) Clearly other factors such as bacterial-dependent gut metabolite production that modify blood metabolites and immune competence may hold the key to ICI efficacy.(25) Although the taxonomy of gut microbiota has been under intense investigation, only a few studies have focused on the respiratory microbiome and its relationship to lung cancer.
Microbial Biomarkers
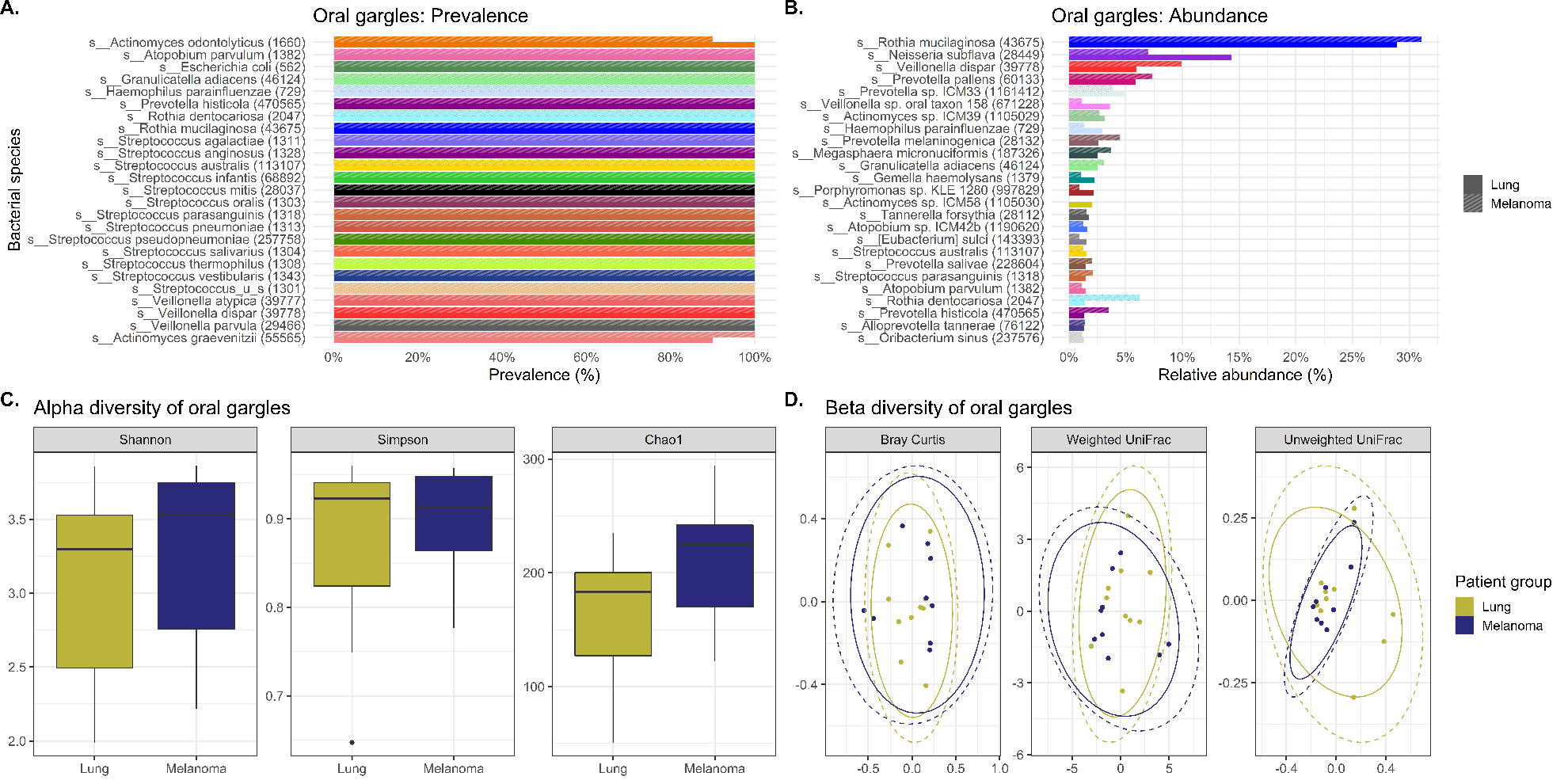
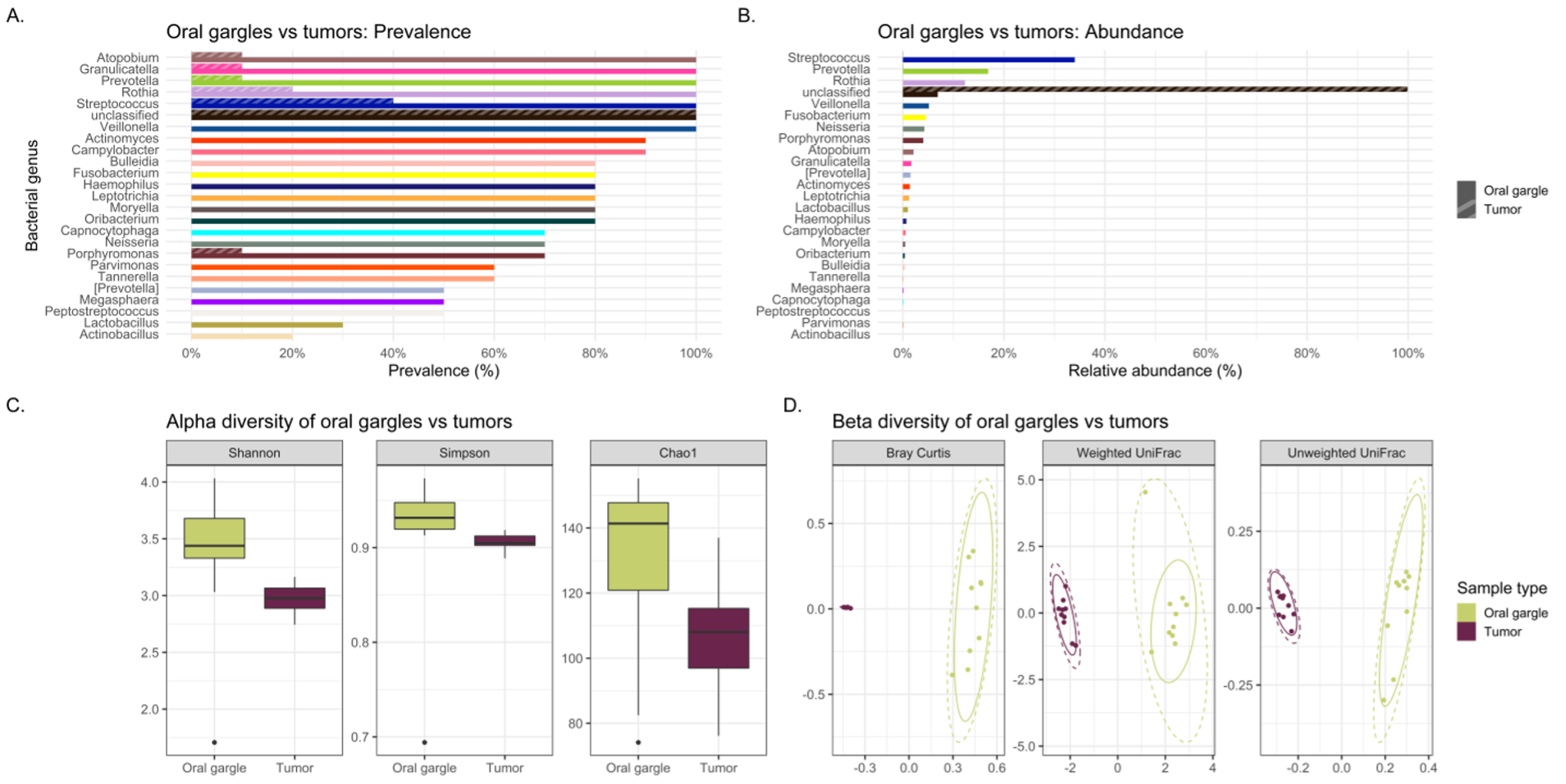
The primary focus of this study was to evaluate the oral and lower airway microbiome compositions of lung cancer cases compared to melanoma controls to reveal differences with potential applications towards biomarker studies. Therefore, we compared tracheobronchial lavages and oral gargles that were collected from both lung cancer cases and controls. The results demonstrate that there are few significant differences in overall microbial composition using prevalence, abundance and diversity measures in the oral gargles between lung cancer and melanoma patients, indicating the readily available, noninvasively sampled oral gargle microbiome would not likely serve as a lung cancer biomarker.
Beta diversity refers to the variation between the samples of one community (group) compared to another community, such that the microbiome composition of one group with a higher beta diversity indicates a greater difference from the other group. By 16S rRNA gene sequencing data, beta diversity measured by Bray Curtis dissimilarity demonstrated significant differences (p=0.022) between case and control lavages indicating that the bacterial communities of lung cancer versus melanoma lavages were distinct, although no such trend was observed for gargles. While the lung cancer and control tracheobronchial lavages were significantly different by 16S rRNA-derived beta diversity, it is difficult to say that the lavages will be able to distinguish lung cancer from non-lung cancer patients since these results were not replicated by WGSS.
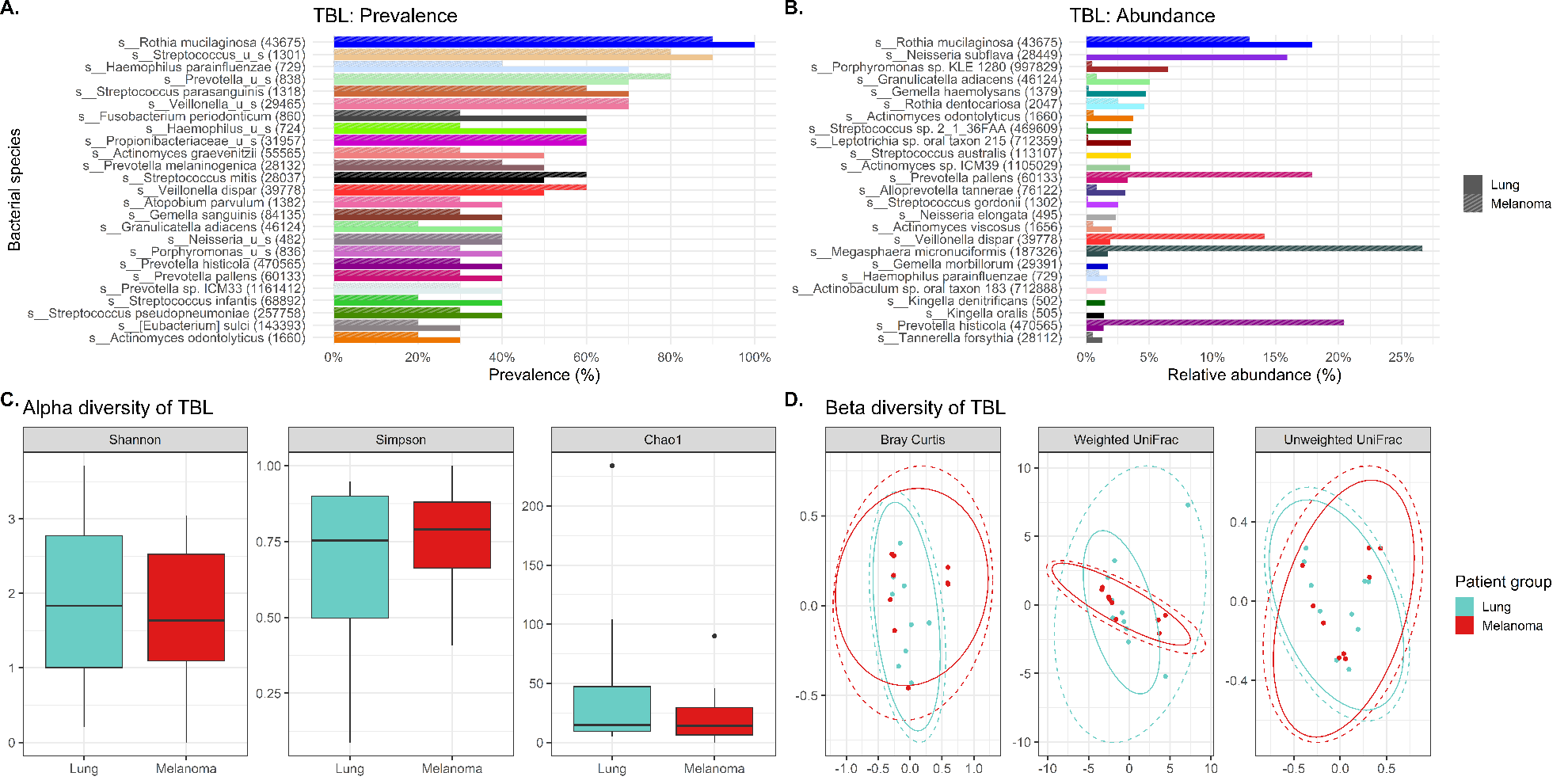
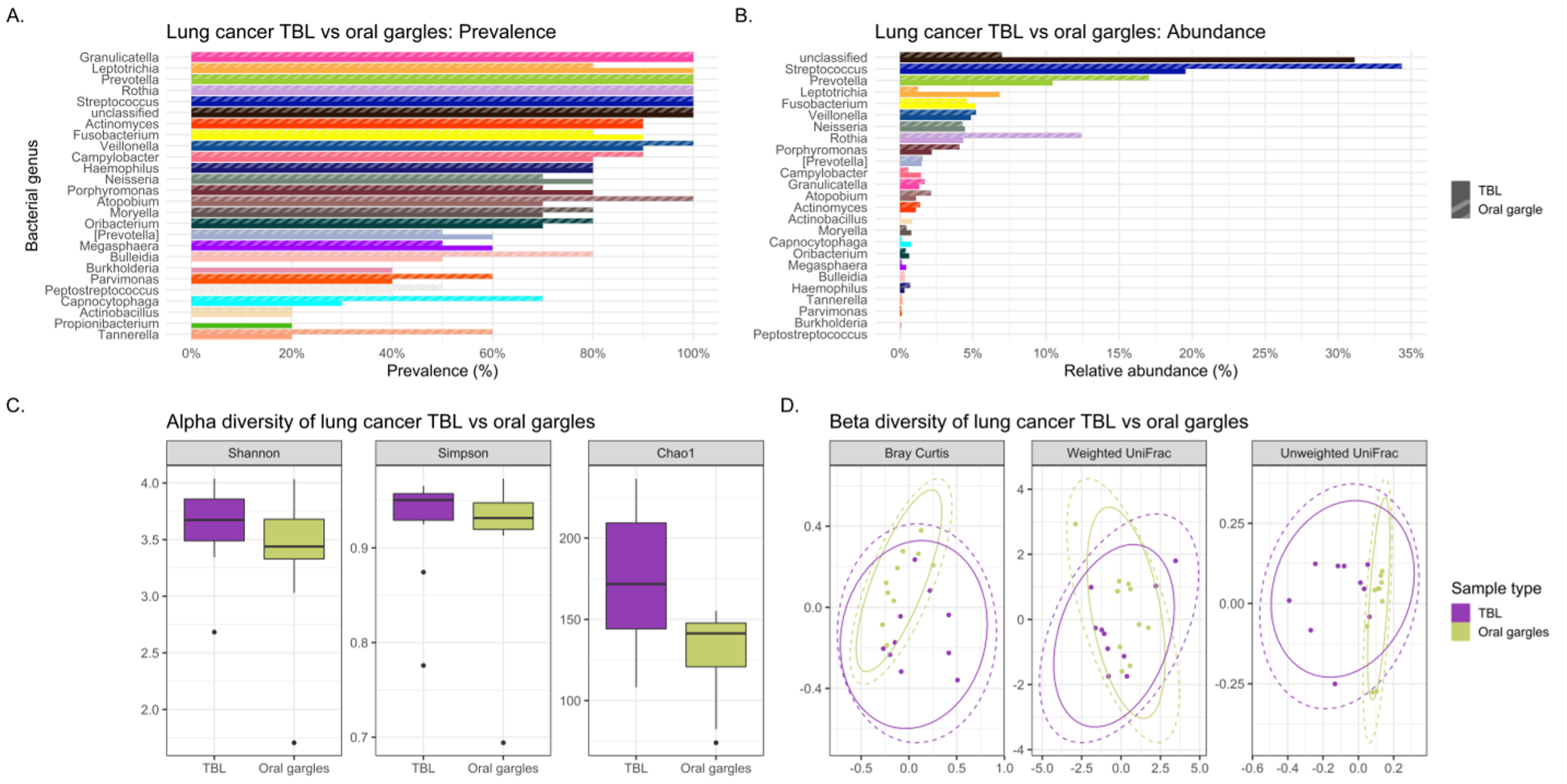
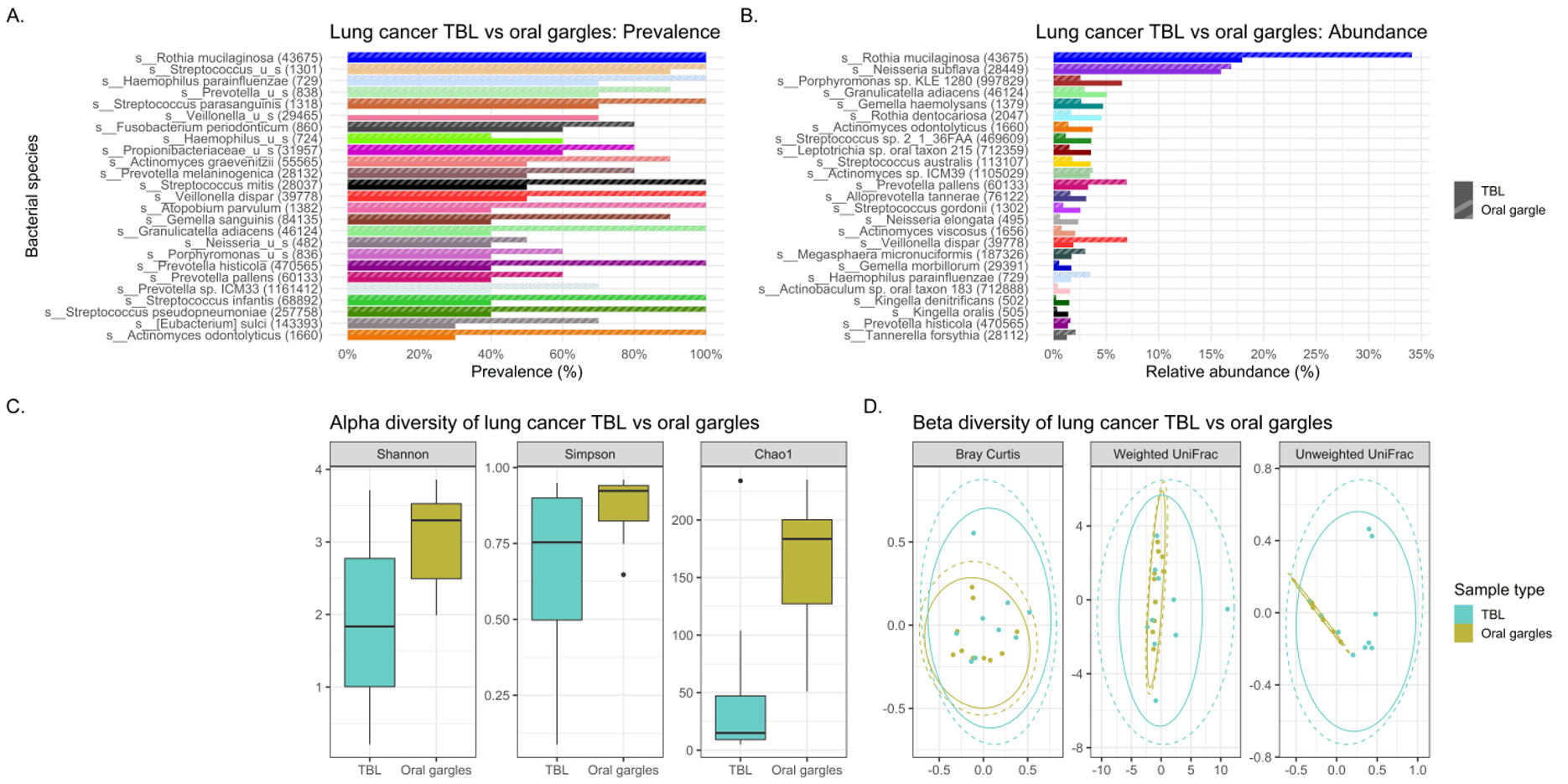
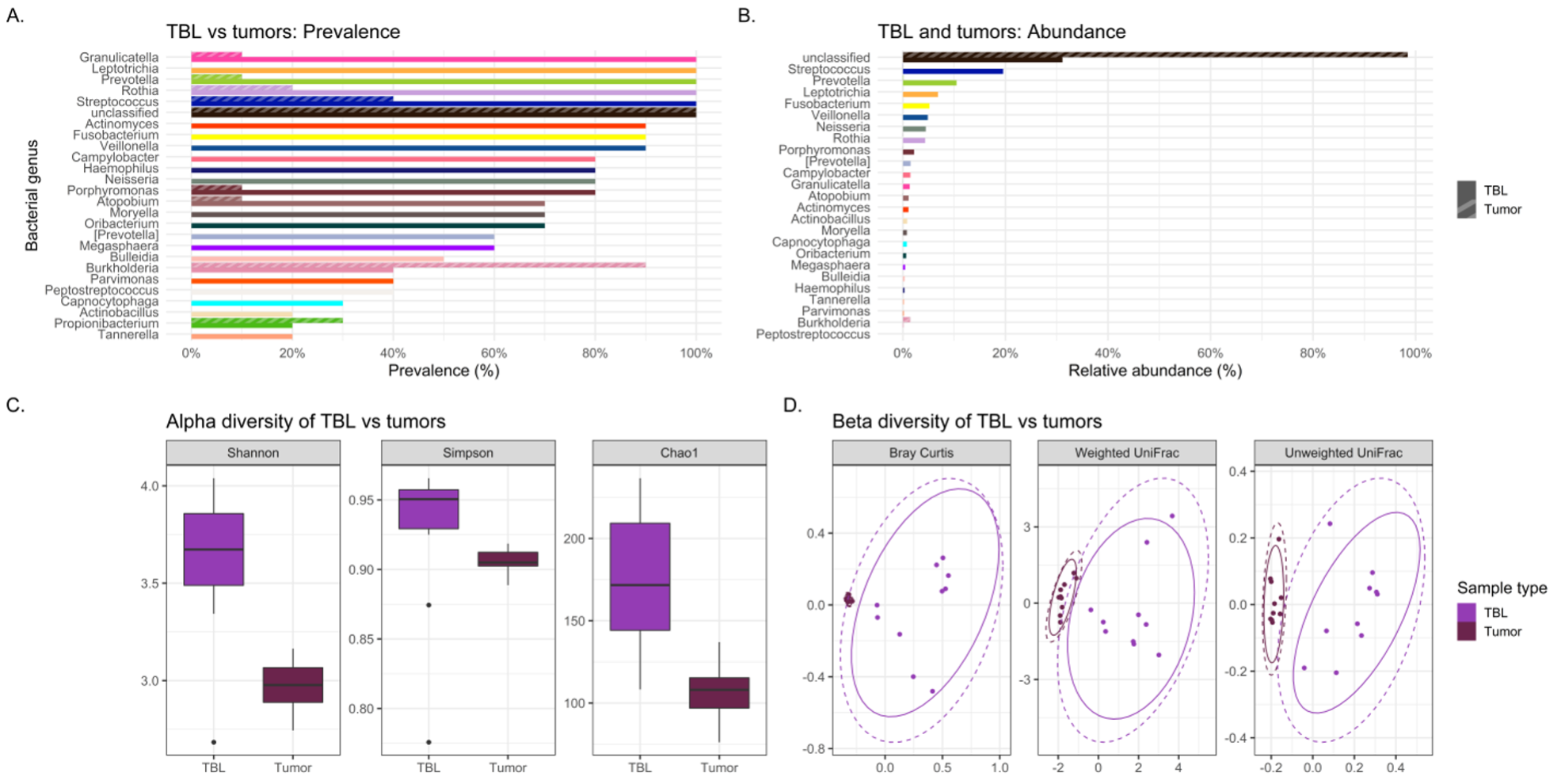
Abundance denotes the percentage that a specific bacterium contributes to a sample’s overall composition. Whereas prevalence refers to the number (percentage) of cases in a specific group in which a bacterium are detected. Interesting trends were observed in abundance and prevalence of the lavages. Most noteworthy was Granulicatella adiacens was more prevalent and abundant in cases. This is a well-recognized oral commensal bacterium that has been etiologically linked to endocarditis.(26) We found this bacterium as one of the top 25 most abundant genera in lung cancer lavages, and it was much higher prevalence appearing in virtually all lung cancer tracheal lavages (100%) versus only some control lavages (30%), despite being similarly abundant in gargle specimens of both groups. Granulicatella adiacens is the same organism that Cameron and associates found in the sputum of lung cancer patients but not controls in a recent pilot study, suggesting this as a potential novel biomarker of lung cancer.(27) Replication of this finding in our study suggests that this microbe may actually be important to further investigate as a potential diagnostic biomarker,(27) and possibly even a predisposing factor to the development of lung cancer.
Additionally, lavage from the lower airways of our lung cancer cases harbored numerous supraglottic bacteria Neisseria (oral commensal), Capnocytophaga (oral commensal), Leptotrichia (oral commensal) and Moryella with twice the prevalence compared to control lavages. Neisseria subflava which commonly colonizes the dorsum of the tongue was also found is high abundance in lung cancer lavages.
LEfSe (linear discriminant analysis effect size) analysis is used to validate biomarkers by detailing features (bacterial taxa in lavages in this case) that distinguish two groups from one another based on relative abundances. In our study, the LEfSe analysis did show several bacterial taxa, including Fusobacteria and Neisseria (especially the oral commensal N. subflava) to be significantly 8-fold differentially abundant in the tracheobronchial lavages of lung versus melanoma patients. These intriguing results strongly support continued research into the tracheal microbiota as potential biomarkers of lung cancer, especially the highly prevalent and abundant Granulicatella adiacens and Neisseria subflava.
Our study also investigated the potential utility of the oral gargle or tracheobronchial lavage microbiomes as proxies for the tumor microbiome in lung cancer. If the lavage and oral microbiomes were similar to the tumor microbiome, these less invasive sample types could be utilized to study the tumor microbiome more easily. Initially, lavages and gargles were compared to see if the gargle could potentially mimic the lavage microbiota. However, significant differences were found between both bacterial and viral community structures (i.e., beta diversity) and alpha diversity in lavages and gargles. That is, the gargle microbiota were dissimilar from the lavages and cannot be used as a representation of the lavage microbiota.
Alpha diversity refers to the variation (how diverse it is) of bacteria within a single sample, with a higher alpha diversity usually associated with a more diverse, healthier microbiome. In our study, the alpha diversity of lavages versus gargles was likewise different, with gargles consistently maintaining higher bacterial and viral diversity by WGSS. LEfSe, performed on both 16S rRNA gene sequencing and WGSS data, also showed many differentially abundant bacterial taxa and some viral taxa between lavages and gargles. Unfortunately, as a result these differences prevent oral gargles from acting as clinical proxies for tracheobronchial lavages. Further differences were identified between the tumor, gargles and lavages that preclude using these sample types as proxies of one another. This was not surprising, however, considering previous literature that has identified significant differences between lung tissue and oral microbiomes.(7)
Despite these results, two recent studies have revealed the prognostic biomarker potential of the lung microbiome: one identified associations of the bronchoalveolar lavage microbiome with recurrence,(28) and another identified Enterobacter in this same sample type associated with worse survival,(19) emphasizing the importance of continued investigation of the lung microbiome in lung cancer. It has already been hypothesized that Enterobacteriaceae, a bacterial family which express the common antigen lipopolysaccharide and identified in our study to be significantly more abundant in lavages and tumor tissue versus oral gargles in lung cancer cases, may induce inflammation in lung cancer that could be associated with poor prognosis.(19) Other studies have suggested that some microbiota may opportunistically invade damaged lung epithelium, caused by smoking, and drive tumorigenesis through production of free radicals like ROS/RNS that can damage the TP53 gene.(20) Mouse models further suggest that lung microbiota may contribute to γδ-T cell activation, which are cells that go on to release the cytokines IL-17A and IL-22.(29) These cytokines appeared to co-occur with tumor progression in the mice.(29) Additional studies are needed to provide substantiated evidence of the mechanistic relationships between the microbiome, the immune system, and lung cancer.
Microbiome of Tumor and Non-Neoplastic Lung
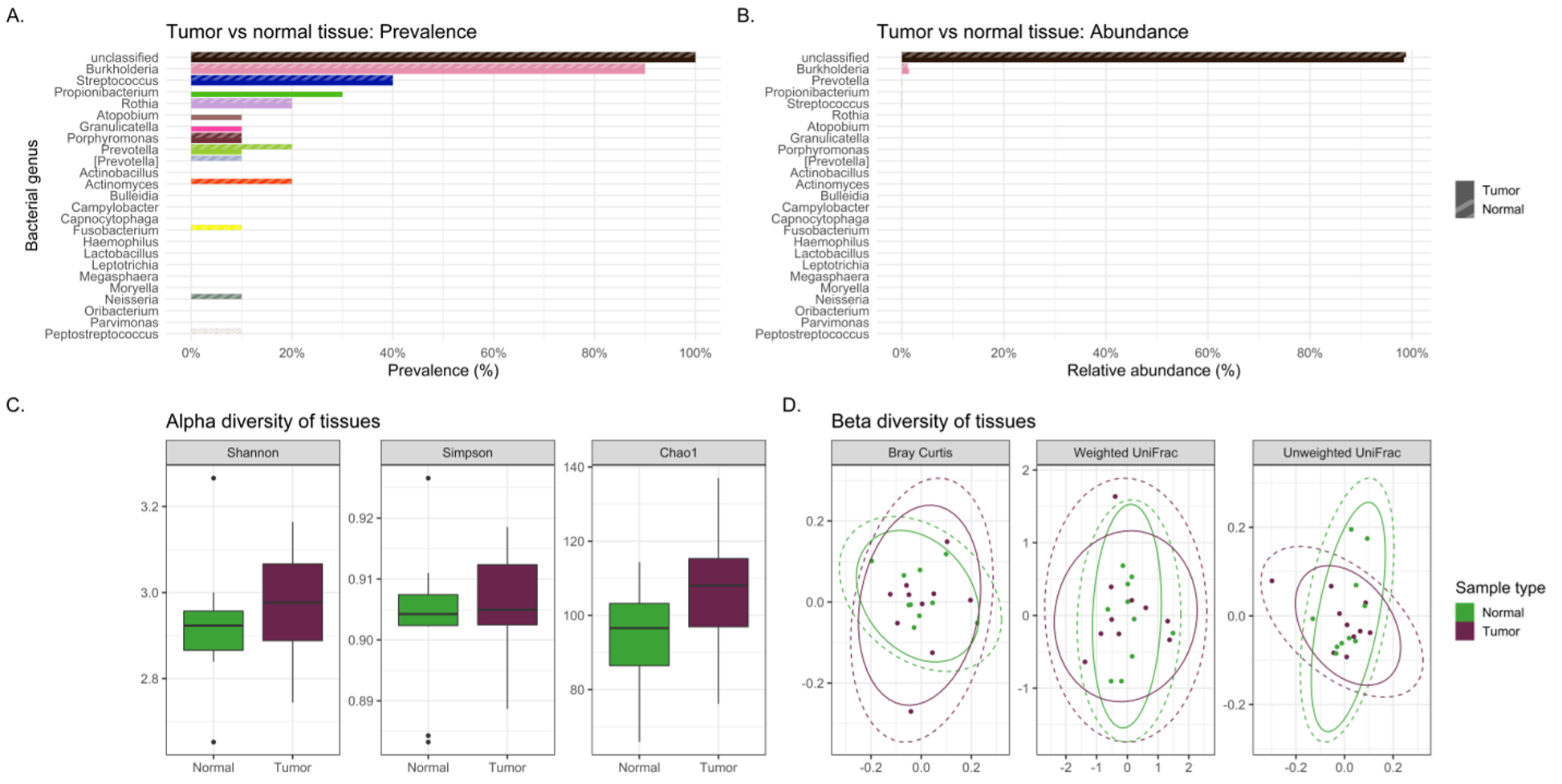
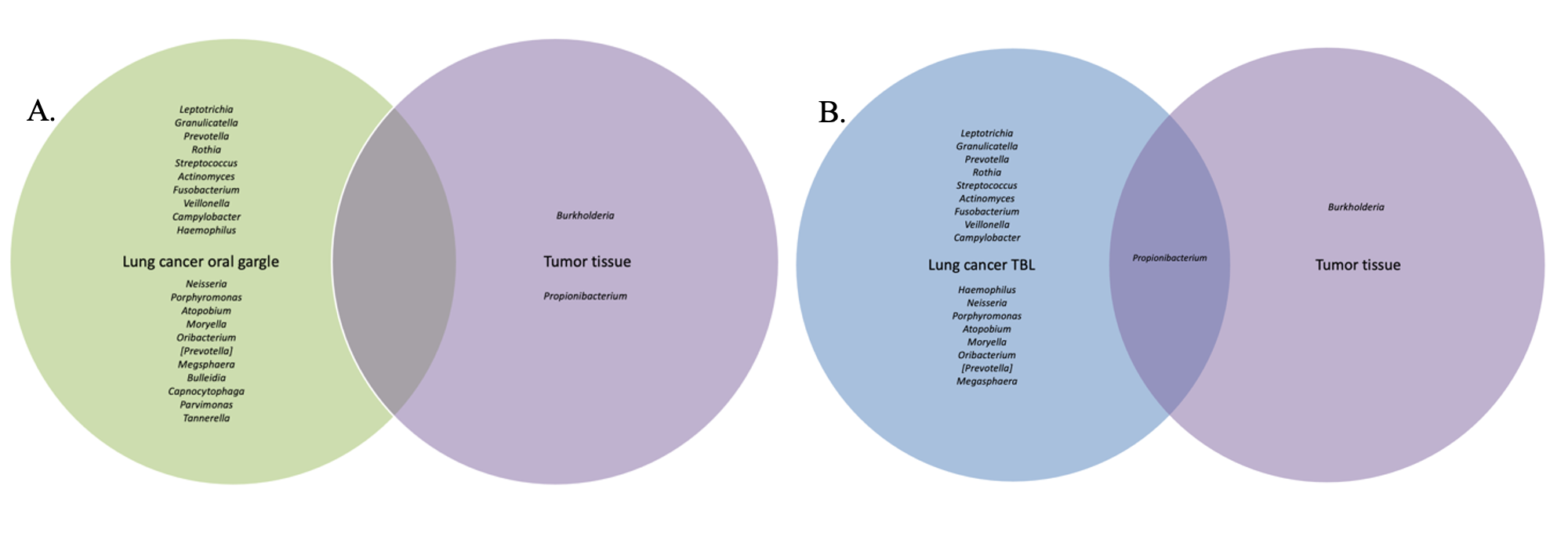
Differences in the composition of the tumor and normal non-neoplastic tissue microbiomes of lung cancer patients were examined to highlight differences that might suggest a microbial contribution to lung carcinogenesis. If the microbiome signatures differed slightly but maintained somewhat similar microbial signatures between tumor and normal tissue, it may indicate certain microbes from the normal lung environment that could have contributed to tumorigenesis, or at least were opportunistic inhabitants of the tumor microenvironment. Indeed, sequencing revealed no significant differences in bacterial relative abundance, alpha or beta diversity between tumor and normal tissue samples. Interestingly, normal tissue had lower alpha diversity compared to tumor tissue, contrary to that observed between tumor and healthy tissue controls previously.(30) Finally, slight variations in bacterial prevalence were identified: higher prevalence of the genera Granulicatella and Burkholderia in tumors was observed, as well as higher prevalence of Neisseria and Fusobacterium in normal tissues.
The genus Granulicatella in particular has been found in a previous study to inhabit the tumor microenvironment, and as it becomes increasingly anaerobic there is production of useful metabolites for this genus.(9) In the current study, Granulicatella was also identified in higher prevalence not only in tumor and normal tissue but also was more prevalent in tracheobronchial lavages of lung cancer patients versus melanoma controls. Hosgood and associates also found a strong correlation between the finding of Granulicatella enriched in the oral and sputum samples of lung cancer patients compared to controls.(31) This provides some intriguing preliminary data suggesting a possible carcinogenic role for some bacteria or at least opportunistic inhabitants of the tumor microenvironment, but testing in larger cohort studies is needed.
Overall, tracheal lavages and gargles do not appear to provide a consistent microbial signature for the tumor microbiome either. In fact, significant differences were observed between the lavage and tumor microbiomes. By all three alpha diversity indices, lavages maintained higher bacterial diversity than tumor tissue and, by all three beta diversity indices, bacterial communities are different between lavages and tumor tissue. LEfSe revealed a large number of bacterial genera more abundant in lavages, like Granulicatella and Neisseria, but one genus was more abundant in tumor tissue, namely Burkholderia, an important Gram-negative pathogen of lung infections in cystic fibrosis patients(32) and is the causative agent in the life-threatening respiratory illness meliodosis.(33) Similarly, beta diversity indicated significantly different bacterial community structures between oral gargles and tumor tissue. This was not surprising considering previous literature that has identified significant differences between lung tissue and oral microbiomes.(7) Indices of alpha diversity also showed gargles to be significantly more diverse than tumor tissues, and LEfSe revealed the genus Burkholderia to once again be more abundant in tumor tissue versus oral gargles.
Overall, lavages and gargles cannot accurately stand in as proxies of the tumor microbiome given the substantial differences between them. However, the genus Burkholderia, in particular, appeared more abundant and prevalent in tumor tissue versus both lavages and gargles, suggesting a potential role in tumorigenesis or at least opportunistic inhabitance of the tumor microenvironment.
Microaspiration
Microaspiration is a common event, occurring in as many as 50% of healthy people,(34) although it is unknown how many have persistent colonization of the tracheobronchial tree with oral commensals. Previous studies by Segal and associates(35) demonstrated enrichment of oral commensals in the lower airways of normal individuals is associated with increased host inflammatory tone and increase in checkpoint inhibitor markers. This lower airway dysbiotic signature was found by Tsay and colleagues to distinguish between patients with lung cancer and benign lung nodules.(36)
Particularly notable differences in our study are the marked 16-, 6- and 6-fold higher abundance of the oral commensals Neisseria subflava, Granulicatella adiacens and Leptotrichia in the lung cancer lavages versus controls. Also, the dysbiotic tracheal microbiome had extensive 2-3 times enrichment of oral microbiota (Granulicatella, Capnocytophaga, Leptotrichia and Neisseria) in lung cancer patients compared to controls (Table 2), perhaps contributing to an inflammatory environment. The control lavages have a markedly reduced abundance of oral taxa, suggesting microaspiration and inflammation occurs to a larger extent in lung cancer patients compared to the control lavages. Indeed, beta diversity studies revealed significant differences (p=0.022) in bacterial community structures between the lung cancer and the control melanoma lavages.
Patnaik and associates also found oral aspiration as the source of lower airway microbiota in lung cancer with the actual microbial community in bronchial lavage correlating with the recurrence of lung cancer after resection.(28) Tsay and colleagues, using RNA-seq analysis of lower airway samples, found that the supraglottic predominant taxa in the trachea were associated with upregulation of inflammatory pathways for p53 mutation, PI3K/PTEN, ERK and IL6/IL8, such that enrichment of the lower airway with oral commensals may increase local immune tone with upregulation of IL1, IL6, and ERK/MARK, in turn promoting tumor progression, hence suggesting microaspiration may be involved in lung cancer pathogenesis.(36)
Microvirome
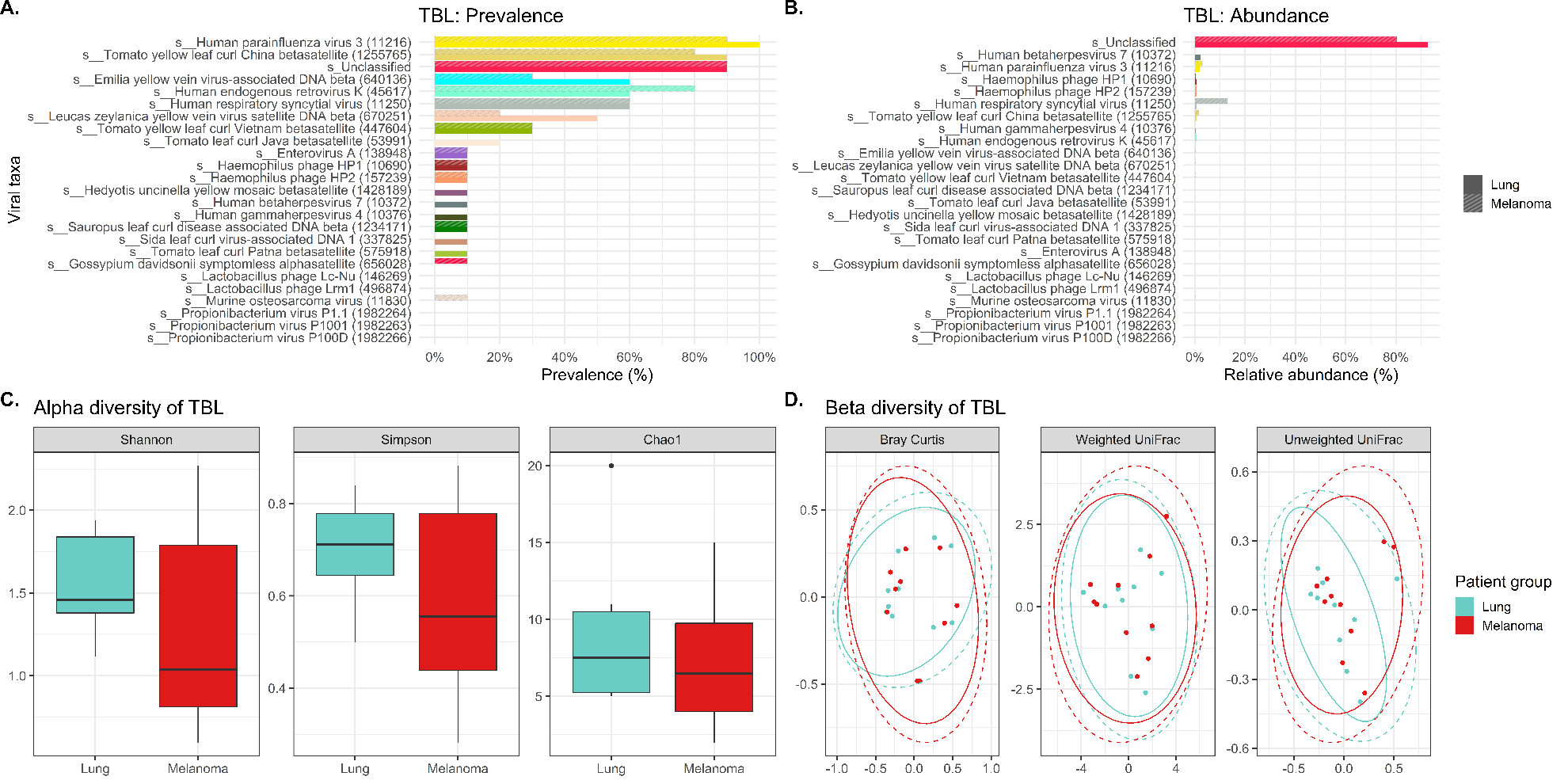
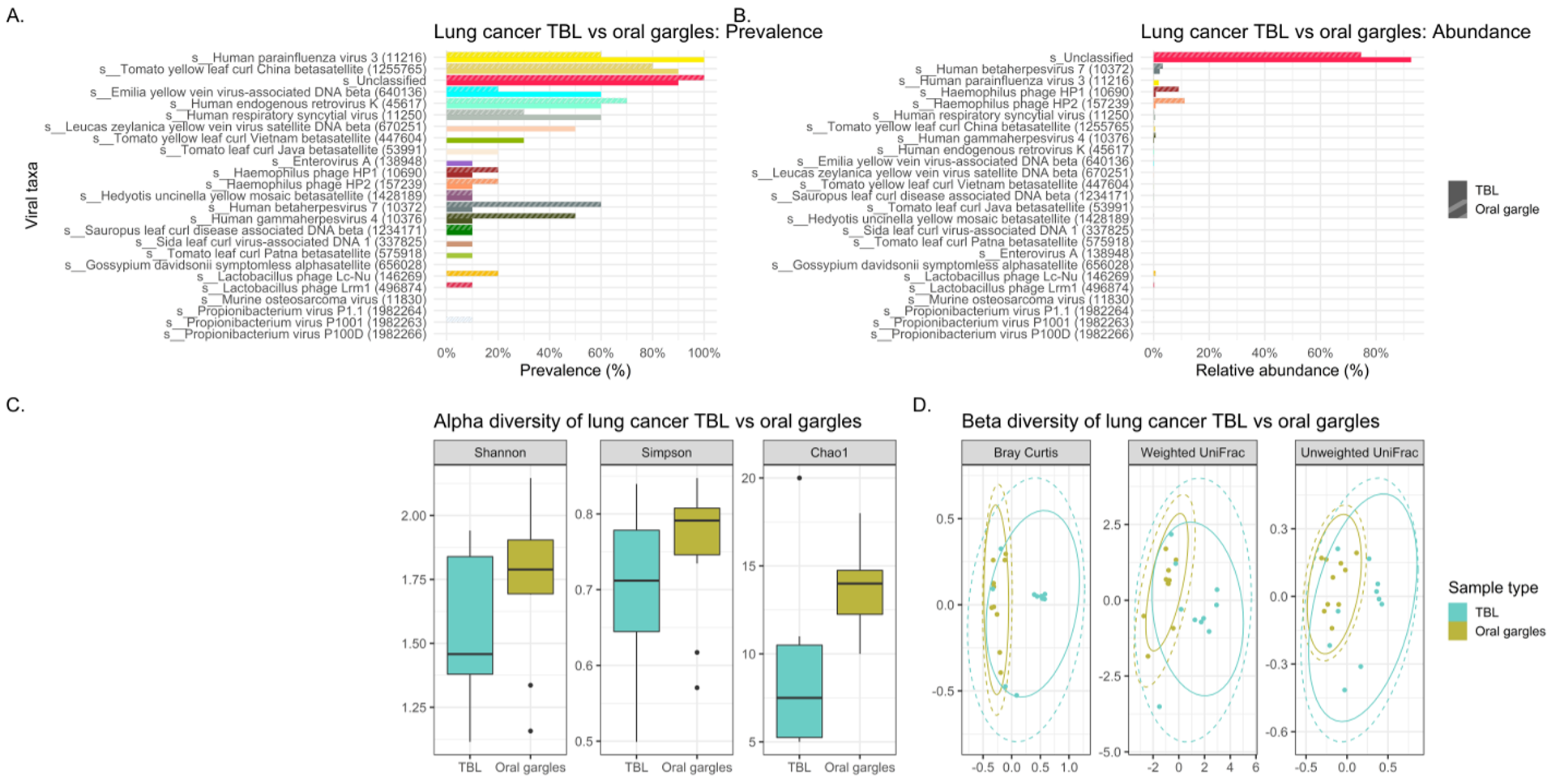
While the characterization of the bacterial (and fungal) members of the human microbiome including the respiratory tree has blossomed in the last decade, studies of the viral component are incomplete. Many challenges exist with exploration of the microvirome due to its low biomass. Even the use of high throughput sequencing technologies is hampered by the small fraction of total DNA in the sample often present in concentrations too low (< 1% of the reads) to be detected without amplification.(37) Additionally, there is the difficulty with the contaminating human and bacterial DNA and RNA in samples. Finally, the vast majority of reads with WGSS are commonly called “viral dark matter” since the results can’t be annotated into taxonomic categories due to lack of species available in databases.(37) As a result, only a relatively small number of viruses were identified in our study likely because of the low abundance and inability to classify common viral organisms.
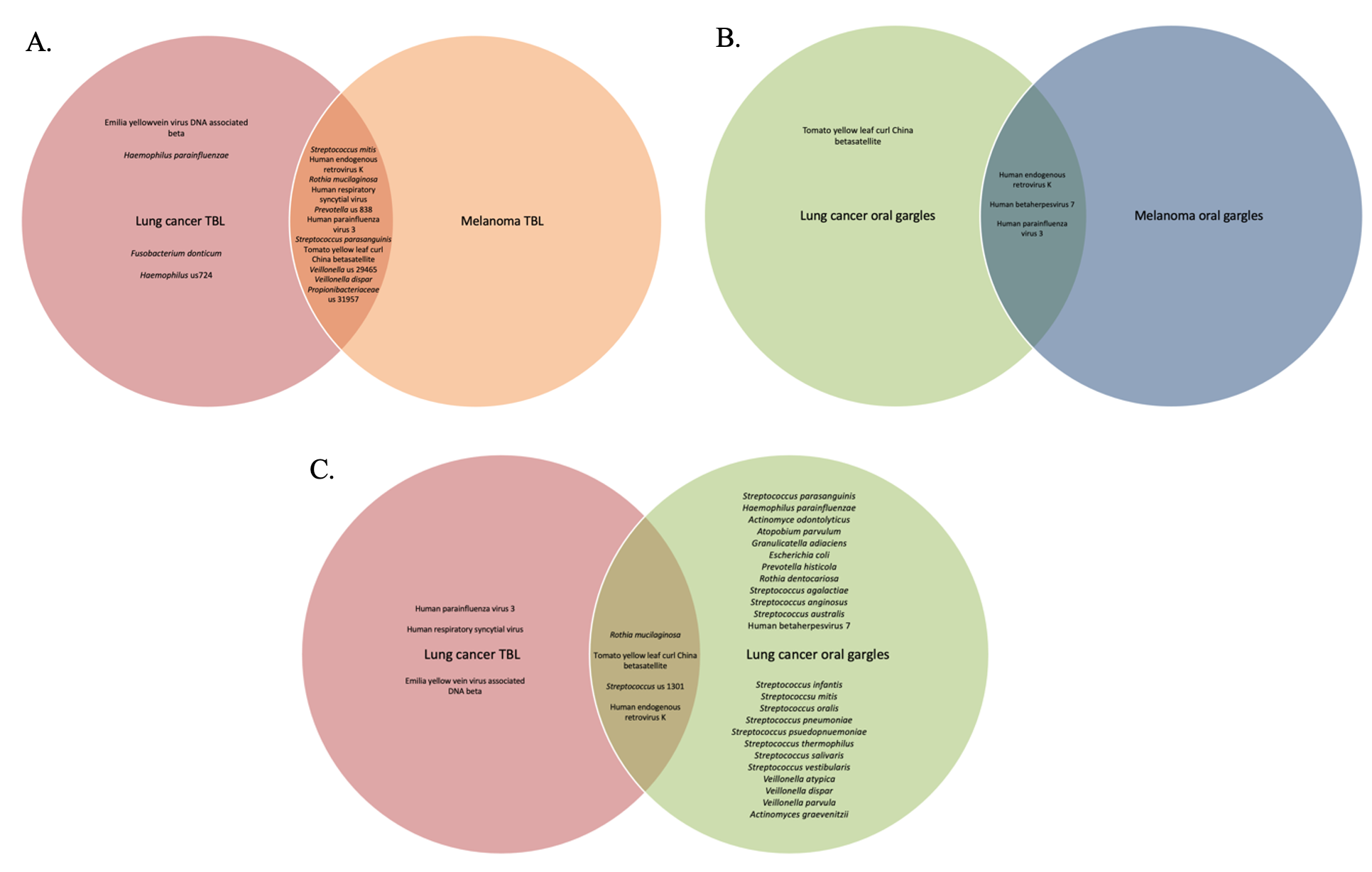
The virome of the tracheal lavages on our study was assessed through WGSS sequencing identifying largely similar prevalence and relative abundance between cases and controls in terms of both lavages and gargles. There was a higher abundance of human respiratory syncytial virus in the melanoma versus lung cancer patient lavages, and conversely human betaherpesvirus 7 was more abundant in lung cancer lavages versus controls. Oddly, many of the more prevalent viruses we identified in both cases and controls are plant pathogens (e.g., yellow vein viruses and tomato yellow leaf curl viruses), although yellow leaf curl is known to infect tobacco plants, which could possibly enter the respiratory tree by cigarette smoking.
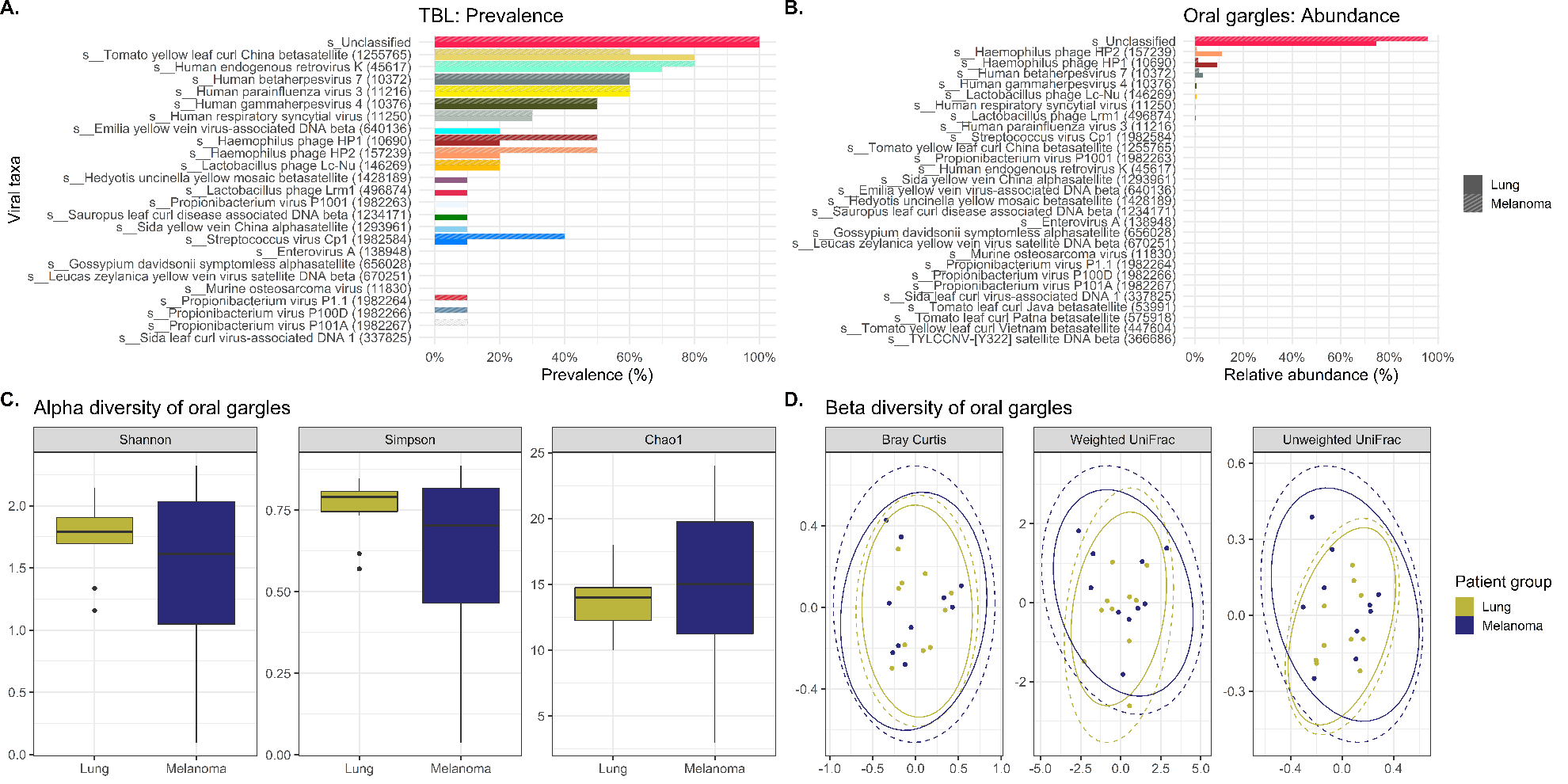
Viral signatures in the oral gargles demonstrated bacteriophages targeting Haemophilus bacteria were more prevalent in melanoma controls versus lung cancer cases. Human-tropic viruses, such as endogenous retrovirus K and betaherpesvirus 7, were similarly prevalent between cases and controls. Although LEfSe identified several unclassified viral signatures as being significantly different between cases and controls, neither alpha nor beta diversity indices demonstrated any significant differences between cases and controls. However, unclassified viral signatures were the most highly abundant in cases and controls.
Comparison of viral signatures between the oral gargle and lavages in the cases demonstrated the prevalence appears different between these two sample types. The most prevalent viral signature in lavages was human parainfluenza virus 3 and the human respiratory syncytial virus compared to gargles. However, human gammaherpesvirus 4 was identified more commonly in gargles. LEfSe revealed several viral taxa significantly differentially abundant between gargles and lavages. Alpha diversity for viral signatures was higher across all three indices in gargles compared to lavages. Finally, beta diversity revealed quite significant differences in viral community structure between gargles and lavages.
Unfortunately, our WGSS and bioinformatics approaches left the vast majority of the viral taxa unclassified in lung cancer lavages (92.6%) and in gargles (74.6%), thus hampering meaningful evaluation of the microbiome.
LIMITATIONS AND STRENGTHS
The small sample size of this study results in significant limitations that may have obscured statistically significant differences in microbiome compositions between lung cancer cases and melanoma controls. In addition, the small subsample sizes prevent us from appropriate sub-analysis of smokers versus non-smoker results. Future research will require larger cohorts to allow sufficient power to detect clinically meaningful differences that could hold biomarker potential. Additionally, a more thorough evaluation of contamination should be implemented in future studies, though our case and control samples were processed in the same way such that contamination should theoretically not result in substantial difference in microbiome signatures between our comparison groups.
Due to the study’s case-control design, the effect of changes in the microbiome over time could not be established to identify when microbial alterations may have occurred in lung cancer patients as compared to the controls. Therefore, further research into microbial dysbiosis in lung cancer will ideally require collecting samples at various time points using prospective cohort designs. This will enable a better understanding of when microbial dysbiosis occurs and how it is associated with clinically important events, such as disease initiation, progression, or treatment response.
Despite these limitations, this study has several major strengths including the direct comparison of the oral, tracheal, lung tumor and non-neoplastic lung microbiome versus the oral and tracheal microbiome of control patients without lung cancer. Also important is the use of WGSS in addition to 16S rRNA gene sequencing of the specimens. WGSS enabled greater taxonomic resolution, specifically to the species level—more so than 16S rRNA gene sequencing would have enabled alone.
WGSS additionally enabled elucidation of viral, not merely bacterial, signatures to generate a more holistic view of the microbial environments among the different sample types. However, since the vast majority (93%) of viral signatures were unclassified, we are conducting additional research studies focusing on the more specific PCR approaches targeting specific viral taxa suspected to be associated with lung cancer, including human retroviruses, human papillomavirus,(38) and hepatitis B virus,(39) demonstrated in our prior pan-microbial array study of biobanked frozen lung cancers.(40)
Ultimately the question arises as to why does a dysbiotic, inflammatory tracheobronchial microbiome appear to be uniformly associated with lung cancer, and perhaps the answer lies in the multifactorial nature of carcinogenesis as suggested by the human papillomavirus (HPV) and cervical cancer picture. HPV has been convincingly proven to cause 99.7% of cervical cancer.(41) If a woman is found to have high risk HPV on her pelvic exam, then she is at elevated risk for the malignancy, yet at most only 8% of high risk HPV-positive women ever develop either pre-cancerous cervical changes or frank cancer.(42) Recent studies suggest that the primary factor determining the ability of HPV to transform cervical cells is the vaginal microbiota, such that a dysbiotic, inflammatory microbiome is needed. An eubiotic, low diversity, low pH vaginal microbiome, particularly dominated by lactobacillus species, likely help clear HPV infections and are also cytotoxic by secreting bacteriocins that modulate the immune system to inhibit viral activity. However, the dysbiotic, proinflammatory microbiome induces oxidative DNA damage and promotes viral transformation of the cervix by the resident HPV.(43)
Therefore, we might postulate a similar scenario for the consistent finding of a dysbiotic tracheobronchial microbiome in lung cancer patients. If some or all lung cancer is “caused’ by one or more oncogenic viruses such as HPV,(44) bovine leukemia virus,(40) and HTLV-1(40, 45), then the development of a dysbiotic, inflammatory tracheobronchial microbiome, such as that found in the current study and others, may be the promoting factor that allows existing colonized, oncogenic viruses to cause malignant transformation in the lung. However, this attractive hypothesis will require a number of future studies to substantiate.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}