The process of genome reannotation combined with detailed manual reviewing encompasses the re-identification and labeling of characteristic features of a sequenced genome, and is a process that has been performed extensively for numerous organisms, including bacteria [16, 17]. Unlike the human genome, which is about 1.3% protein coding, 90% of the bacterial genome codes for proteins, with only short intergenic stretches [18]. Precise genomic annotation is thus fundamental to the further interpretation of the biochemical and physiological characteristics of organisms, to provide detailed information on protein coding sequences, pseudogenes, non-coding RNAs, repeat sequences and various other genomic data [19]. In this study, we reannotated the genome of the E. coli strain ER2566 through a reannotation pipeline, as illustrated in Figure 1, with high speed and accuracy.
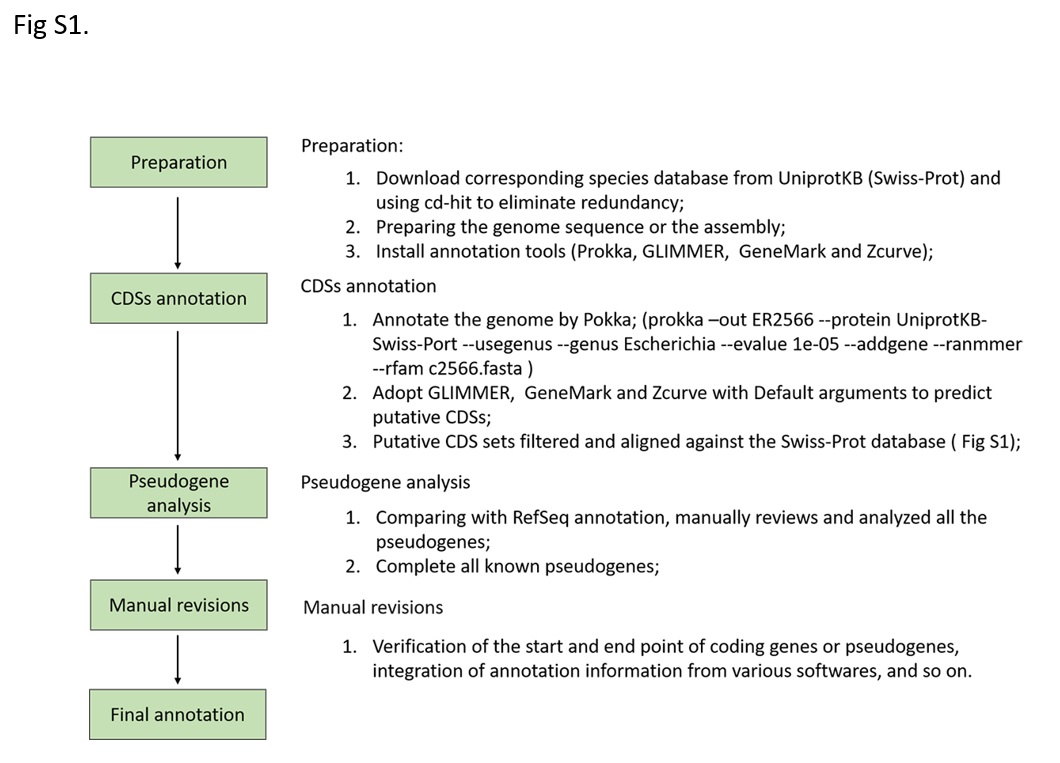
We employed a series of automated annotation tools combined with manual inspection to reannotate the ER2566 genome (Fig S1). In our pipeline, the automation part using Prokka combined with others gene finders (GLIMMER, Zcurve and GeneMark) could finish a complete bacterial genome annotation in about 30 min. Compared with some online tools, this pipeline showed higher speed and accuracy. For example, NCBI provides a Prokaryotic Genomes Annotation Pipeline service via email, with a turn-around time of several days [7]. RAST is another web server for annotating bacterial and archaeal genomes that provides results within one day [20]. Some local stand-alone annotation tools, such as RATT [21], Rapid Annotation Transfer Tool, can transfer annotations from a high-quality reference to a new genome on the basis of conserved synteny. However, due to the limitation of its algorithm, RATT cannot effectively identify pseudogenes, indels, etc. Besides, numerous automated tools have been developed for genome annotation, including Mypro [22], MAKER [23], BlastLKOLA [24] and so on. To avoid false-positive predictions, the algorithms of these annotation tools are designed to balance specificity and sensitivity of their results [25]. In contrast, combination of multiple ab initio gene finders combined with BLAST searching and manual inspection will help to confirm identified genes and generate more accurate annotations.
Improvement in coding sequences (CDS)
For the systematic reannotation of the CDS, the prediction and identification of coding genes occurred in two stages (Fig S2). In the first stage, Prodigal software was used to predict a total of 4,180 CDSs on the complete ER2566 genome deposited in GenBank (accession number NC_CP014268.2). Using sequence alignment to the Swiss-Prot database [26] by Blastp [27], with a threshold e-value of <10-6, all CDSs were annotated to provide accurate information regarding the sequences and functions of the enrolled proteins. A total of 4,023 (96.2%) of the 4,180 CDSs were annotated as protein-coding genes, with the remaining CDSs (136 CDSs; 3.3%) marked as hypothetical genes, with no registration in the Swiss-Prot database, and 21 CDSs (0.5%) marked as pseudogenes by manual inspection. To improve upon this prediction, three other well-established gene finders-GLIMMER, Zcurve and GeneMark-were independently used, identifying 4,231, 4,287, and 4,213 CDSs, respectively. These putative CDS sets were subsequently filtered by Blastn against the first Prodigal-predicted CDS set. Overall, an additional 428 CDSs were found: 194 CDSs were identified using GLIMMER, 123 using GeneMark, and 201 using ZCURVE. These additional 428 CDSs were then searched against the Swiss-Prot database by Blastp with a stricter threshold e-value <10-10, coverage >80%, and an identity >70%. This filtered out 402 of these additional CDSs, resulting in an additional 43 genes. This led to a total of 4,066 protein-coding CDSs (4,023 + 43) included in the reannotation of the ER2566 genome, along with 136 CDSs for hypothetical genes.
Due to trimming or splitting, genes with real function can often be incorrectly assigned as pseudogenes through protein homology alignment. In our reannotation, we manually reviewed and analyzed 194 pseudogenes from the RefSeq database annotation. In total, 123 of the 194 pseudogenes were directly identified as coding genes and are now found in the reannotated list. while the remaining pseudogenes without any function had been retained. These newly identified protein-coding genes included 34 mobile genetic elements that encode transposases and had been considered to be important in evolution as a common type of genetic change. For instance, a genomic positive strand region (3,296,328 - 3,297,025 bp), previously annotated as a pseudogene without function, was identified to harbor two genes, insA and insB, which are homologues of the insertion element protein, IS1, and related to DNA binding and transposase activity (Fig. 2a) [28]. In addition, two annotated pseudogenes (C2566-RS05300 and C2566_RS05310) and one hypothetical protein (C2566_RS22600) in the RefSeq annotation (range 1,088,980-1,094,720) were reannotated as three new genes (lacZ1, lacZ2, and ECBD_2906, respectively), and one related pseudogene, C2566_RS05305, was removed; these changes are consistent with previous results [5] (Fig. 2b). These three new genes are flanked by the lactose permease gene lacY upstream and the lactose operon repressor gene lacI downstream, both of which are essential to the lac operon system. This reannotation had uncovered genes related to the transcription and translation of genes encoding the lactose operon repressor in ER2566 strain.
In comparing to RefSeq annotation, a total of 190 protein-coding CDSs were removed, as they were identified as either hypothetical proteins or pseudogenes, with most of them having no assigned function (Additional File 1). Meanwhile, 237 new CDSs were added (Additional File 2). The complete reannotation list can be found in Additional File 3. ER2566 strain is a BL21/K-12 hybrid strain, where about 6% sequence of K-12 strain replaces about 7% sequence in BL21(DE3) genome. The genome alignment of BL21 and ER2566 demonstrates a high degree of consistency [29] (Fig. 3). The recent version of BL21(DE3) annotation contains 4,197 CDSs, in which 3,873 (92.3%) CDSs were identified in ER2566 annotation as identical gene symbols or alias as well. The unidentical 7.7% CDSs annotated in BL21 genome as compared to ER2566 is comprised of ~7% sequence corresponding to the hybrid part in ER2566 and other CDSs that does not have an official gene name (Additional File 4). Overall, we determined 4,197 protein-coding genes for the ER2566 genome, including 136 CDSs labeled as hypothetical proteins and 4,061 CDSs, which 3,873 CDSs identical to BL21(DE3) that account for about 99% of total 3,903 CDSs (4,197*93%) equivalent to BL21(DE3) CDSs within ER2566 genome. This reannotation effectively eliminated the possibility of false interpretations introduced by the original annotation and provides a more integral view of the regulatory networks in ER2566 strain (Table 1).
Table 1: Overview of the differences between the original annotation, the reannotation and BL21(DE3) annotation
|
Original annotation
(NZ_CP014268.2)
|
Reannotation
|
BL21(DE3)
|
|
Genome length
|
4,478,958 bp
|
4,558,953 bp
|
|
Plasmids
|
None
|
|
G+C%
|
50.81%
|
50.83%
|
|
Genes(total)
|
4,364
|
4,627
|
4,440
|
|
Protein_coding genes
|
4,054
|
4,202
|
4,197
|
|
Pseudo Genes
|
194
|
71
|
70
|
|
tRNAs
|
85
|
87
|
85
|
|
rRNAs
|
22
|
22
|
22
|
|
Miscellaneous RNAsa
|
9
|
245
|
66
|
|
Backbone genes
|
4,170 (4,054 protein-coding genes,85 tRNA genes,22rRNAs and 9 misc RNAs)
|
4,556 (4,202 protein-coding genes,87 tRNA genes,22rRNAs and 245 misc RNAs)
|
4,370 (4,197 protein-coding genes, 85 tRNA genes,22rRNAs and 66 misc RNAs)
|
a : The concept of miscellaneous RNA includes ncRNA, tmRNA and all other ncRNAs.
Integrated proteogenomics search database (iPtgxDB) is widely used to provide protein expression evidence and could confirm the validity of the annotation, which was used to identify the short protein-coding genes that have numerous functions [30]. Thus, the combination of transcriptome data and reannotation results was used to generate an integrated proteomics database, which provided an important optimal basis for genome-scale regulatory or metabolic predictions and comprehensive exploration on the genome information and underlying gene functions (Additional File 5).
Miscellaneous RNAs improvement
Miscellaneous RNAs, such as transfer RNA (tRNA), ribosomal RNA (rRNA), and other non-coding RNAs (ncRNA), play pivotal biological roles in cellular activity. To date, about 119 RNAs molecules in the E. coli ER2566 strain have been identified, including 85 tRNAs, 22 rRNAs, and 12 ncRNAs [6]. However, almost all of these ncRNAs are missing from the original RefSeq annotation. We used Aragorn 1.2, Barrnap 0.9, and infernal 1.1 ncRNA finders independently to predict genes coding for tRNAs, rRNAs, and ncRNAs. Compared with the auto-annotation, 2 tRNAs and 230 ncRNAs are new, with most having functions in translation, DNA replication, and expression regulation (Additional File 6). In addition, most of the ncRNAs functions have been verified experimentally. For instance, about 94 of the nucleoid-associated ncRNAs molecules play key functions in DNA-RNA interactions [31]. Meanwhile, fragments per kilobase of transcript per million mapped reads (FPKM) value was used to quantify the transcription level of the newly added ncRNAs and to analyze the transcriptomics data of ER2566 under different induction conditions (Additional File 7). Quantitative analysis indicates that 85% (208/245) of newly added ncRNAs are detectable, while the other 15% (37/245) ncRNAs are undetectable possibly due to some specific functions requiring certain conditions. In summary, our reannotation introduced 230 new ncRNAs and 2 new tRNAs, with an overall tally of genes encoding for 87 tRNAs, 22 rRNAs and 245 ncRNAs.
Variant calling of whole-genome re-sequencing under consecutive subculture
Genomic variations in bacterial species usually reflect an evolutionary response that occurs under various external—usually unfavorable—environmental stressors. Thus, we next performed variant calling to identify any nucleotide-level differences (i.e., single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and/or structural variations) in the ER2566 strain. There are two approaches for variant calling: by mapping reads against the reference genome directly or by assembling a de novo genome to compare against a reference genome. In most cases, mapping reads produces a better resolution for SNPs and indels than genome assembly, whereas the latter is optimal for identifying structural variants and regions with high divergence. Here, we used both methods to interrogate the ER2566 genome (Fig. 4).
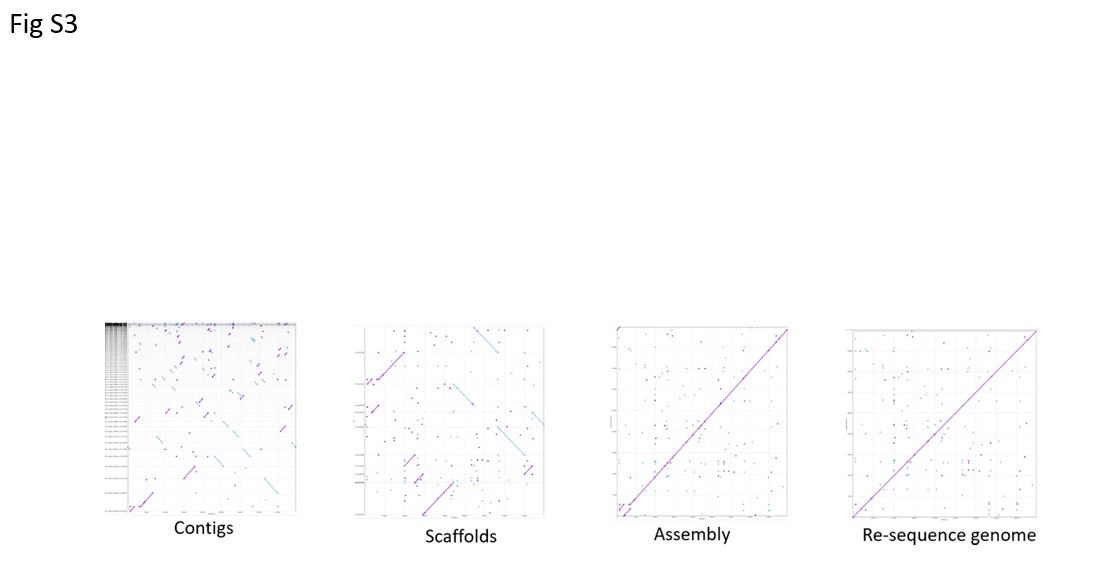
First, we re-sequenced the whole genome of the ER2566 strain grown in our laboratory under consecutive subculturing. Two different-sized insert libraries (500 bp, 2,000 bp) were built, and a total of 10.0 million paired-end reads of 90 bp in length were generated using an Illumina HiSeq 2000. The raw reads were mapped to the C2566 reference genome (NC_CP014268.2) with a good coverage depth (>100-times). No SNP or indel was detected. A pipeline optimized for longer assembly was designed to accomplish the re-sequencing of our ER2566 strain. Various de novo assembly softwares were used to construct a confident and long (4,469,460-bp) scaffold, and assembly results for each step were assessed by alignment of final sequences back to the reference genome (Fig S3). One inversion was found (Fig S3c), which turned out to be a genome assembly issue caused by high repetition region and was corrected by subsequent Sanger sequencing. The technical difference between short-read sequencing and single molecule long-read sequencing may result in the generation of inverted region. Nevertheless, our pipeline, by the combination of read mapping, de novo assembly and Sanger confirmation, generated an intact ER2566 genome in our practice. The resequencing of ER2566 also suggested that the continuous subculture of E. coli ER2566 strain in our lab did not cause mutation in the genomic sequence.
Mutation detections by RNA-sequencing under overexpression
RNA-seq is widely used in quantitative gene expression studies for the identification of non-annotated transcripts and polymorphisms, and for RNA editing in transcribed regions. Thus, to identify any variations in the ER2566 genome due to overexpression pressure, we used RNA-seq to analyze the transcriptomes of the ER2566 strain growing at 37°C without plasmids (B37, three replicates) or overexpressing human papillomavirus 16 type capsid protein L1 via plasmid-based inducible expression (Y37, three replicates) (Fig. 5a). From a total of 75.4 million 125-bp paired-end reads, 73.9 million reads (98%) were mapped to the reference genome (NC_CP014268.2) in B37 (control) samples. Yet, for the Y37 (overexpressed) samples, among the 76. 0 million reads, only 29.6 million (39%) reads mapped to the reference genome, which was significantly lower than that for the B37 samples. The cause of lower mapping rates in Y37 samples was due to the large number of mRNAs transcribed by the engineered plasmids which is not related to genome sequence (Table 2).
Table 2:Statistical analysis of RNA-seq data
|
Sample
|
Run
|
Raw sequences reads
|
Unidentified readsa
|
HPV16L1 readsb
|
E. coli readsb
|
|
B37
|
1
|
22,781,394 (100%)
|
250,596 (1%)
|
0 (0%)
|
22,334,273 (99%)
|
|
2
|
22,763,848 (100%)
|
318,694 (1%)
|
0 (0%)
|
22,312,577 (99%)
|
|
3
|
29,884,996 (100%)
|
597,700 (2%)
|
0 (0%)
|
29,276,302 (98%)
|
|
Y37
|
1
|
28,292,578 (100%)
|
4,526,812 (16%)
|
12,731,660 (45%)
|
11,034,106 (39%)
|
|
2
|
25,214,878 (100%)
|
4,286,528 (17%)
|
11,346,696 (45%)
|
9,581,654 (38%)
|
|
3
|
22,521,416 (100%)
|
3,378,212 (15%)
|
10,134,638 (45%)
|
9,008,566 (40%)
|
a: Three biological replicates of each samples were analyzed. unidentified RNA-seq reads could include unidentified nucleotides (Ns), short reads, low quality reads, unaligned recombinant protein reads and a larger number of mRNA from plasmids. b: HPV16L1 reads and E. coli reads respectively represent Human papillomavirus and Escherichia coli organism reads.
The variant detection analysis using BactSNP revealed one mutation in the Y37 samples (1,094,824 position; Fig. 5b), located in the non-coding region 19-bp downstream from the lacI gene. Interestingly, lacI is the highest transcribed gene in the Y37 samples by comparing the FPKM for all genes (Additional File 8). The analysis of transcriptome data indicated a mutation of C to T substitution at position 109,824 in the three replicates of Y37 samples, as confirmed by nearly 100% mutation rate in all observed reads (910,914 Out 911,345 reads). Furthermore, the mutation was found in three replicates of B37 samples as well, with a mutation rate up to 85% (655 Out 773 reads). Surprisingly, such mutation could not be detected in the bacterial genome by Sanger sequencing (Fig. 5c). The discrepancy between transcribed RNA and genome sequence may arise from the modification during RNA transcription instead of a sequencing error. The presumption is supported by the mutation position being located in a high-efficiency RNA methylation site, which is often accompanied with spontaneous deamination of 5-methylcytosine and consequently producing thymine in aqueous solution [32]. It is interesting to clarify whether the overexpression of lacI could lead to the increase of RNA methylation rate. The function of this site will be further investigated in our future work, using a combination of gene editing, RNA methylation analysis, and other relevant techniques.
{kind=link}
{kind=link}
{kind=link}