Plant materials and growth conditions.
Euryale ferox HL and its parents, North Gordon Euryale (E. ferox, WT) and South Gordon Euryale (E. ferox, SE), were used as plant materials in this study. Seeds of WT were originally obtained in 2001 from the wild (Gaoyou lake of Jiangsu province) and cultured in the experimental field of Jiangsu Seed and Seed Breeding Base, and seeds of SE were originally obtained from the wild (Suzhou of Jiangsu province) and also grown in the experimental field of Jiangsu Seed and Seed Breeding Base who provided permission for their use in this scientific research. HL (containing 3 lines) were constructed from WT as the female crossed with SE to generate F1 hybrid in Gaoyou (E119°30’, N32°58’), Jiangsu province, China in 2015; later, F1 was selected and self-pollinated to generate F2 in Nanzha (E119°10’, N33°18’), Jiangsu province, China in 2016; and F2 individuals were used to produce F3 by self-fertilization in Gaoyou (E119°30’, N32°58’), Jiangsu province, China in 2017; finally, F3 generation and both of its parents were cultured in the experimental field of Jiangsu Seed and Seed Breeding Base (Gaoyou, Jiangsu province). Collection of all samples completely complies with national and local legislation permission, and no specific permission was required for collecting these plants. The plant materials were formal identified by Professor Qinan Wu, and a voucher specimen of this material has been deposited in Nanjing University of Chines Medicine. HL and the WT were sown in a completely randomized block design with three replications in April 22, 2018. Each plot consisted of 160 individual lines, each separated 2 m from its neighboring lines. The two lines were selected in this study to measure phenotypic traits and conduct transcriptome analyses. The young fruits selected for sampling at the designated development stages were collected and stored at − 80 °C for RNA-seq and RT-qPCR analysis. Seeds were collected at maturity on September 9, 2018; they were selected with 10 replicates for yield traits; additionally, each sample had at least three biological replications to minimize systematic errors.
IAA quantification.
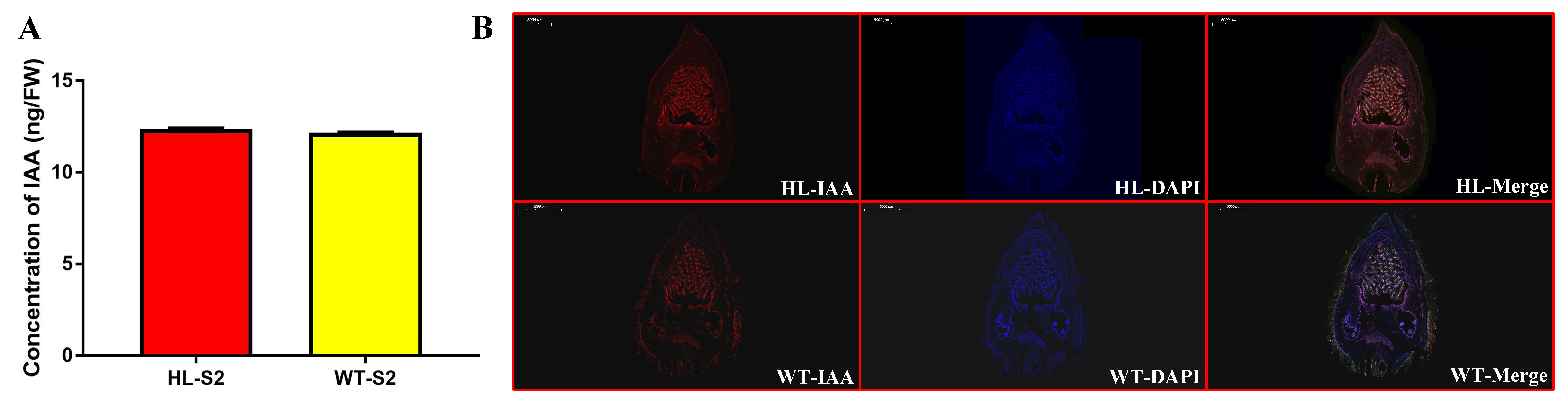
IAA was quantified by liquid chromatography-mass spectrometry (LC-MS), essentially as described[55]. Briefly, 50 mg of frozen tissue was homogenized in 500 μL of extraction buffer (isopropanol: double distilled water: acetic acid= 2:1:0.002, vol/vol/vol). After shaking on ice for 30 min, 1 mL dichloromethane was added to each sample and shaken for another 30 min. Then, samples were centrifuged at 13,000 g for 5 min at 4 °C; the lower phase was dried under a stream of nitrogen gas, and redissolved with methyl alcohol. Each sample solution was injected into the reverse-phase C18 HPLC column (2.1mm × 100 mm, 2.7 μm, Agilent, USA) for LC-MS (Shimadzu LC-30 ultrahigh performance liquid chromatography system, Shimadzu, Japan; AB Sciex QTRAP 5500, SCIEX, USA) analysis. An IAA standard (≥98%, 45533, Sigma, USA) was used as the external standard.
Immunofluorescence staining.
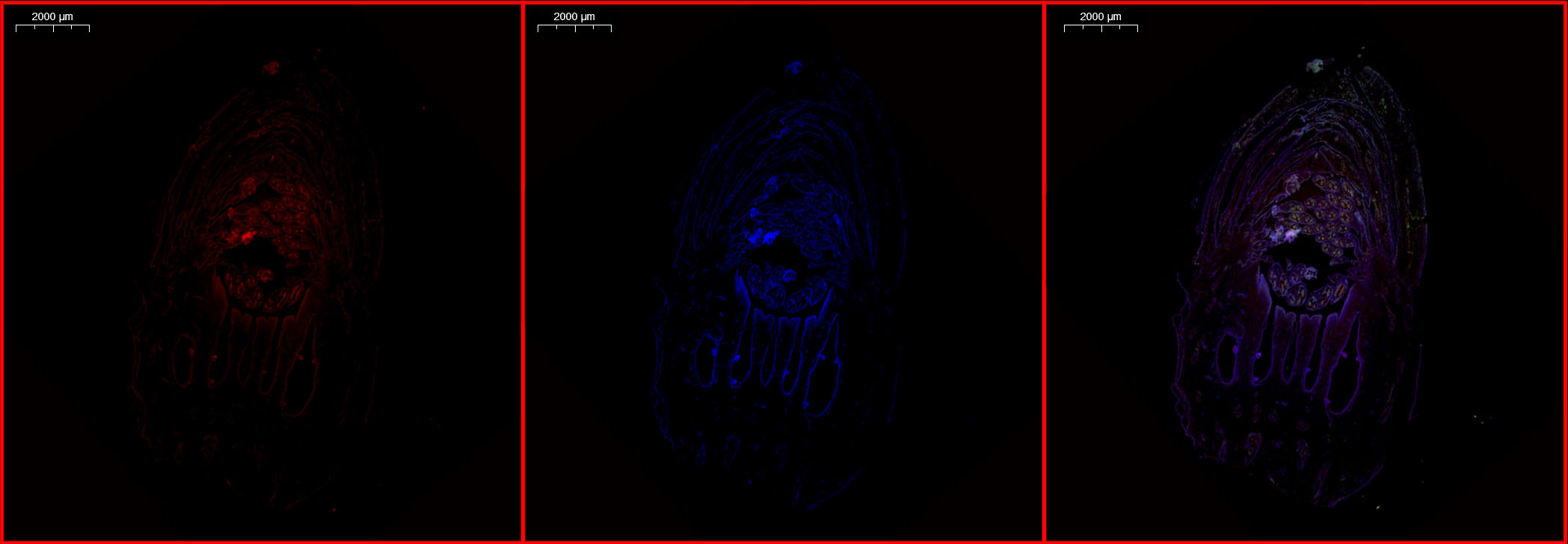
Immunofluorescence staining for IAA was performed on sections of fruits as follows: Briefly, 60 μm fruit sections were cut with a sliding cryotome CM1950 (Leica Microsystems, Germany) and mounted on slides. Sections were dehydrated for 5 min in ascending and descending 25, 50, 75, and 100% ethanol solutions, washed three times for 10 min in phosphate buffer saline (PBS) buffer containing 0.1% (v/v) Tween 20. Sections were pre-treated with 5% (w/v) bovine serum albumin in PBS for 1 h at room temperature to reduce non-specific binding before incubating overnight with anti-IAA antibody (1:200, AS09 421, Agrisera, Sweden) at 4 ℃. After washing three times with 0.1% (v/v) Tween 20 in PBS buffer for 10 min, sections were incubated with Alexa Fluor 594 (1:200, conjugated Goat Anti-Rabbit IgG (H+L), SA00006-4, Proteintech Group, USA) as a secondary antibody for 1 h in darkness at room temperature. Samples were washed three times for 10 min with 0.1% (v/v) Tween 20 in PBS. Fluorescence signals were detected on a Carl Zeiss Axio Scope fluorescence microscope (Gottingen, Germany), and quantitative analysis was performed using ImageJ 1.46. PBS was added instead of primary antibodies as a negative control; we did not detect any unspecific binding of the secondary antibody using this method.
RNA extraction, cDNA library preparation and sequencing.
Before total RNA was extracted from samples, 12 frozen tissue samples were fully grinded separately under liquid nitrogen. RNA quality was determined by Agilent 2100 BisAnalyzer (J06-02, Agilent, USA), and samples with both RIN≥7 and were included for RNA-seq analysis. RNA concentration was determined using a Qubit RNA BR Assay Kit (Q10211, Invitrogen, USA).
To construct the cDNA libraries, 5 μg total RNA per sample was used as the template. The NEBNext Ultra RNA Library Prep Kit for Illumina (E7530S, NEB, USA) was used to generate a series of corresponding libraries, and index codes were applied to each sample. Briefly, mRNA was purified from total RNA using oligod (T) beads. RNA fragmentation was carried out at high temperature in the presence of divalent cations in NEBNext First-Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primers and M-MLV Reverse Transcriptase. Second-strand cDNA synthesis was subsequently performed using DNA polymerase I, RNase H, dNTP and buffer. Then, the cDNA fragments were purified with AMPure XP beads, end repaired, poly (A) added, and ligated to Illumina sequencing adapters. Ligation product size was selected by agarose gel electrophoresis, amplified by PCR, and sequenced using Illumina HiSeq TM X10 by Gene Denovo Biotechnology Co. (Beijing, China).
Identification of differentially expressed mRNAs.
Raw reads generated from high-throughput sequencing were treated as follows. First, to remove adapters that were added for reverse transcription and sequencing, sequences with too many unknown bases (>5%) or low-quality bases (>30% of the bases with a quality score ≤ 20) were removed. The remaining clean reads from each sample were then assembled by Trinity3 with de Bruijn graph.
The unigene sequences were annotated using the following public databases: Universal Protein (Uniprot), evolutionary genealogy of genes (Eggnog), Pfam protein domain database (Pfam), signal peptide server (SignalP), Hidden Markov Model-based transmembrane protein database (TMhmm), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO). The unigene sequences were aligned using BLASTx with an e-value of < 10-5, and the annotations were only saved when the “-max_target_seq” was “1”.
Differential expression was estimated and tested using the RSEM12 software with the EM method. We quantified gene expression levels in terms of Fragments Per Kilo bases per Million (FPKM), calculated the false discovery rate (FDR), and estimated the fold change (FC) and log2 values of FC. Transcripts that exhibited an FDR ≤ 0.01 and an estimated absolute │log2(FC) │≥ 1 were considered to be significantly differentially expressed.
Quantitative real-time PCR (RT-qPCR) analysis.
RNA was extracted using TRIzol (T9424, Sigma, USA) and 1 μg used for cDNA synthesis with the PrimeScript RT Master Mix kit (RR036A, TaKaRa, China). RT-qPCR was performed with a QuanStudio 3 instrument (Applied Biosystems, USA), using 5 μL cDNA, NovoStart SYBR qPCR SuperMix Plus (E096-01B, Novoprotein, China), and 0.2 μL each of two gene-specific primers (Table S1) in a final volume of 20 μL. The thermocycling regime consisted of 2 min at 50 °C, 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 1 min at 55 °C, and 30 s at 72 °C. Disassociation curves verified amplification of a single product. The relative expression of each gene was analyzed using the comparative Ct (ΔΔCt) method with β-actin as a reference gene for normalization. The reference gene was selected according to previous studies[56]. Briefly, gene-specific primers were designed by Primer Premier 5 and checked by melting curve after amplification. The expression stability of each candidate reference gene was compared and ranked by BestKeeper software according to the threshold cycle (Ct) values. Reference genes with a standard deviation value below 1 were considered as stably expressed in this study, and a smaller coefficient of variation indicates a more stable reference gene. Each amplification reaction was performed in triplicate.
Data analysis.
All data were expressed as the mean ± standard error of the mean. Differences between two groups were analyzed using t-test and P<0.05 was considered to indicate a statistically significant difference. Statistical analysis was performed with GraphPad Prism 6.0 software (GraphPad Software, Inc.).
{kind=link}
{kind=link}