All custom software can be found at https://gitlab.com/buchserlab/FIVTools
Cell Culture and Transfection
Human osteosarcoma (U2OS, ATCC HTB-96) cell lines were maintained in McCoy's 5A Modified Medium (16600082, Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS) (16000044, Gibco). Human embryonic kidney (HEK) 293T cells (CRL-11268, ATCC) were cultured in Dulbecco's Modified Eagle's Medium (11965-092, Gibco, Gaithersburg, MD, USA) supplemented with 10% FBS (16000044, Gibco, Gaithersburg, MD, USA), 1% Penicillin-Streptomycin (15140122, Gibco) and 1% non-essential amino acids (11140050, Gibco).
All cell lines were maintained in T75 tissue culture flasks in an incubator at 37°C, 5% CO2 and they were observed daily for growth and overall health. Once confluent, cells were passaged using 0.25% Trypsin-EDTA 1x (25200056 Gibco, Gaithersburg, MD, USA) at a sub-cultivation ratio of 1:10. Live cell counting was performed with the BioRad TC20 automated cell counter. Centrifugation of cell cultures was performed at 1200 rpm for 3 minutes. Lentiviral infection was performed in T75 flasks when cells were 85% confluent. STR profiling, to confirm cell type, was performed using NGS-based analysis by the Genome Engineering and iPSC Center (GEiC) at Washington University in St. Louis. Testing for mycoplasma was performed bi-annually. For all experiments in this paper, either 100x100 or 200x200 micron quad reservoir plates containing 48,000 (12,000 cells per quad) and 36,000 cells (9,000 cells per quad), respectively were used. Prior to plating, microraft plates were prepared by rinsing with 1mL PBS 3 times with 3-minute incubation periods. Cells were added in 200µl media to aid in distribution, then plated and incubated overnight (14-16 hours).
Virus Production and MFN2 Single Mutant Line Creation
MFN2 lentiviral expression plasmids were cloned into the CCIV lentiviral plasmid with a GFP marker 39. In preparation for lentiviral packaging, 8.0 x 105 HEK293T cells were plated into each well of a six well plate and incubated at 37°C overnight. The cells were then transfected with TransIT Lenti-transfection reagent (MIR 6600, Mirus Bio, Madison, WI, USA) using an envelope plasmid (pVSVg: Addgene plasmid # 8454), a packaging plasmid (psPAX2: Addgene plasmid # 12260), and each individual MFN2 expression plasmid in a mass ratio of 0.5/1/0.5 respectively for a total of 2µg. After 48 hours, media was collected, centrifuged, and sterile filtered before being concentrated (Lenti-X Concentrator 631232 Takara Bio, Kusatsu, Shiga, Japan). Concentrated virus was resuspended in 200µL 1xPBS per well, collected, and stored at -80°C.
To create stable MFN2-mutant expressing lines, T75 flasks containing
6 million U2OS cells were infected with 70µL of concentrated lentivirus at an MOI>1 and polybrene was added (NC9840454 Santa Cruz Biotechnology, Texas) at a final concentration of 10µg/mL. They were then incubated for 24 hours, after which the virus-containing media was removed and replaced with fresh, virus-free media. Cells were taken to the Washington University Siteman Flow Core for fluorescent sorting on the Sony Synergy HAPS1, 100-micron sorter. Cells were sorted based on viability and GFP expression (since no puromycin selection was performed, the fluorescent signal from the GFP in the MFN2 plasmid was used to determine transgene expression). GFP expression levels were compared within and across generated cell lines to ensure population purity and comparable fluorescent expression levels. The PRIMPOL KO U2OS cell line was received from the Vindigni lab and was produced by the GEiC.
CRISPR/Cas9 gRNA Library Infection and Induction
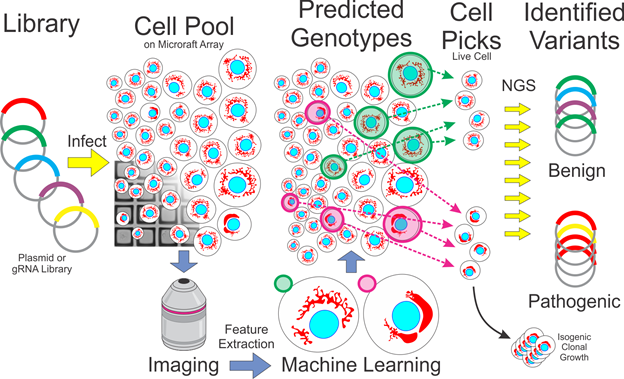
A dox-inducible Cas9 (iCas9) U2OS cell line was generated via CRISPR-mediated homology directed repair. The Cas9 protein, gRNA, and donor construct were introduced via nucleofection. Isogenic iCas9 clones were isolated using the Cell MicroSystems CellRaft Air System and then propagated for further experiments. Presence of the construct was validated via junction PCR 47 prior to propagation. Puromycin-resistant MFN2 scanning gRNA libraries were generated and cloned by the Washington University GEiC. Lentivirus was produced (see Virus Production above) and used to infect iCas9 U2OS cells at an MOI of <0.2 followed by 8µg/mL puromycin selection for seven days. The cells were then allowed to grow in fresh media. At 60-70% confluency, Doxycycline (Cat#: D9891-1G, Millipore Sigma) was added at a final concentration of 2µg/mL. The cells were incubated at 37°C for 48-60 hours before proceeding with staining and imaging.
Staining and Microscopy
The following vital dyes were used; DNA labeling/nuclei (Hoechst, Thermo Fisher H3570), mitochondria (MitoTracker Deep Red, Thermo Fisher M2246), and mitochondrial membrane potential (Tetramethyl Rhodamine methyl ester TMRM, Thermo Fisher I34361). MitoTracker and TMRM were incubated for 40 minutes at concentrations of 0.5 and 1µM respectively. Hoechst was incubated for 15 minutes at a concentration of 10µg/mL (16.2µM). Each plate was rinsed twice with culture media prior to imaging. Images were captured using a 20x 0.45 NA objective in the Cytiva INCell 6500HS Confocal microscope. Exposure times for Hoechst (405 nm) and TMRM (561 nm) averaged 0.15 seconds while MitoTracker Deep Red (642 nm) averaged 0.05 seconds. Confocality was used in the 405 and 642 wavelengths to decrease the background fluorescence of the CytoSort raft plate. Each field-of-view overlapped by 12% of their area. Imaging settings were held constant throughout the course of an experiment. Following imaging, an extra 500µL of cell culture media was added to the CytoSort raft plate (additional liquid helps the Cell MicroSystems Air System isolate microrafts).
Image Analysis and Quality Control
Image tracing and feature extraction was performed using Cytiva’s INCarta software. Mitochondrial puncta were identified (within 20µm of the nuclei using the ‘networks’ algorithm) and quantified for each cell as were a set of texture features. Raft coordinates were recorded for each cell (using FIVTools/ CalCheck, included in the GitLab repository). Images were also curated semi-manually (via FIVTools/ CalCheck) to ensure that out-of-focus images were excluded. The cell feature dataset was joined with the image quality data and raft position mapping data described above by custom software (via FIVTools/ main window). Post tracing quality control was performed with each dataset in Tibco Spotfire Analyst. First, aberrant tracing artifacts were excluded based on nuclear area, nuclear form factor, and proximity to the raft’s edge. Next, non-nuclear debris and dead nuclei were excluded by gating on nuclear area, intensity, and cell intensity. Rafts with too many cells (>6) or a fiduciary marker were excluded. This filtered set of cells was used as the input for machine learning downstream.
Machine Learning and Model Generation
After exporting the quality-controlled cell-based feature table, we built a machine learning model that could distinguish pure populations of WT cells from pathogenic mutant cells. The models were trained real-time on the day of the experiment since we were working with live cells which needed to be physically picked within the next few hours to maximize viability. A variety of machine learning platforms (Microsoft AzureML Studio, Tibco Spotfire, Tibco Statistica, and H2O.ai.) and algorithms were employed to predict an unknown cell’s genotype class. Generally, logistic regression was performed in Spotfire and random forests, boosted trees, support vector machines, gradient boosted machines, and artificial neural networks were trained in the other platforms. Models were evaluated on labeled populations that were withheld during training. Based upon model performance on the testing dataset, a model was selected and deployed to the unlabeled cell populations. Starting with the strongest prediction scores, a list of cells with raft locations was generated.
Cell Capture and DNA Extraction
Cells were isolated using the Cell MicroSystems CellRaft Air System. CytoSort raft plates were received from Cell Microsystems (Durham, North Carolina). Given a list of raft coordinates, the Air System used a needle to eject each individual raft and transfer the raft to a semi skirted 96-well PCR plate (1402-9200, USA Scientific) via a magnetic wand. Each well of the PCR plate contained 5µL extraction buffer (molecular grade water with 10mM Tris-HCl (pH 8.0), 2mM EDTA, 200 µg/mL Proteinase K, and 0.2% TritonX-100). Raft isolation was confirmed twice through post ejection imaging of the raft location and through visual inspection using a Leica S8AP0 dissection scope. Genomic DNA was extracted in a thermocycler immediately following raft isolation by incubating at 65°C for 15 minutes then 95°C for 5 minutes.
Single-Cell DNA Amplification
Amplification of single-cell DNA prior to library preparation consists of two separate amplifications. An initial preamplification is conducted using extracted DNA with KOD Hot Start DNA Polymerase (71842-4, Millipore Sigma, Burlington, MA, USA) according to manufacturer’s instructions using all 5µL of extracted DNA in a total reaction volume of 20µL. Pre-amplified product was processed through an AMPure XP (Catalog: A63882, Beckman Coulter, Brea, CA, USA) bead clean up according to the manufacturer’s instructions using 10mM Tris-HCl pH 8.5 as elution buffer. The second amplification uses the cleaned template and BioLine MyTaq HS Red Mix 2x (C755G97,Meridian Life Sciences, Memphis, TN, USA), according to manufacturer’s instructions, including 5% by volume DMSO. Primers in the second amplification contained universal 5’ tags to be compatible with Illumina library preparation (Forward tag: 5′-CACTCTTTCCCTACACGACGCTCTTCCGATCT-3′, Reverse tag: 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′).
For amplification of MFN2 cDNA, primers amplifying the entire cDNA were used in the first amplification step, followed by multiplexed amplification of two specific regions containing the relevant mutations. Genotyping of the RFP-GFP cells used multiplexed primers that amplified specific regions in both the RFP and GFP regions. All primers are listed below in Table 2.
Table 1
|
Name
|
Sequence (Excluding tags, where necessary)
|
PCR Stage
|
|
pMFN2.All.F
|
GCTCTTCTCTCGATGCAACTCT
|
1
|
|
pMFN2.All.R
|
GCAGGTACTGGTGTGTGAAC
|
1
|
|
pMFN2.1.F
|
CACATGGCTGAGGTGAATGC
|
2
|
|
pMFN2.1.R
|
GCAGGAAGCAATTGGTGGTG
|
2
|
|
pMFN2.2.F
|
CTCAGAGTCCACCCTGATGC
|
2
|
|
pMFN2.2.R
|
CACTTGAAAGCCTTCTGCGAG
|
2
|
|
RFP.F
|
GTTCATGCGCTTCAAGGTGC
|
1, 2
|
|
RFP.R
|
CAAGTAGTCGGGGATGTCGG
|
1, 2
|
|
GFP.F
|
TGAAGTTCATCTGCACCACCG
|
1, 2
|
|
GFP.R
|
TCGCCCTCGAACTTCACCTC
|
1, 2
|
|
PRIMPOL.F
|
GCAACCCAGTTTTGAAACCA
|
1, 2
|
|
PRIMPOL.R
|
TCGATGTCCAGCTTTCCTCT
|
1, 2
|
|
gRNA.F
|
CTTGTGGAAAGGACGAAACACC
|
1, 2
|
|
gRNA.R
|
TTGTGGATGAATACTGCCATTTGT
|
1, 2
|
Illumina Library Preparation
These methods are expanded from Connelly et al. and Bell et al. After amplification with universal primers, each plate was amplified with specific forward and reverse Illumina index primers that indicate the PCR plate position and a unique plate ID. PCR amplification was performed with BioLine MyTaq HS Red Mix 2x (C755G97,Meridian Life Sciences, Memphis, TN, USA) according to the manufacturer’s protocol, pooled, and then cleaned using AMPure XP bead (A63882, Beckman Coulter Life Sciences, Indianapolis, IN,USA) cleanup procedure in the original amplification. DNA was quantitated on a NanoDrop One Spectrophotometer (Thermo Scientific, ND-ONE-W) before being submitted to the Center for Genome Sciences and Systems Biology (Washington University) to generate 2x250 reads on the Illumina MiSeq platform.
Sequencing Analysis
Illumina paired reads were demultiplexed by the core facility and FastQ files were returned. The rest of the analysis was performed with laboratory software available on Gitlab (FIVTools/ LA, “Library Aligner”). Reads were joined and trimmed, then aligned with small sequence fragments at the genetic sites of interest containing the sequence to mutant or WT alleles. The result was a ‘counts’ table that gave the number of reads containing each 20-mer for each well. 20-mer search fragments are listed below in Table 3. After accounting for isolation and genomic amplification errors, around 80% of the isolated cells genotypes were captured.
Table 2
|
Name
|
20-mer (relevant mutations/deletions bolded)
|
|
MFN2_V69
|
TGGACCCCGTTACCACAGAA
|
|
MFN2_V69F
|
TGGACCCCTTTACCACAGAA
|
|
MFN2_L76
|
ACAGGTTCTGGACGTCAAAG
|
|
MFN2_L76P
|
ACAGGTTCCGGACGTCAAAG
|
|
MFN2_R94
|
TGCTGGCTCGGAGGCACATG
|
|
MFN2_R94Q
|
TGCTGGCTCAGAGGCACATG
|
|
MFN2_D221
|
CTGGATGCTGATGTGTTTGT
|
|
MFN2_D221=
|
CTGGATGCTGACGTGTTTGT
|
|
MFN2_P251
|
CTCTCCCGGCCAAACATCTT
|
|
MFN2_P251A
|
CTCTCCCGGGCAAACATCTT
|
|
MFN2_R280
|
CATGGAGCGTTGTACCAGCT
|
|
MFN2_R280H
|
CATGGAGCATTGTACCAGCT
|
|
MFN2_W740
|
AAAGCCGGTTGGTTGGACAG
|
|
MFN2_W740S
|
AAAGCCGGTTCGTTGGACAG
|
|
RFP_guide
|
GGCCACGAGTTCGAGATCGA
|
|
RFP_control
|
AAGGTGCGGATGGAGGGCAG
|
|
GFP_guide
|
TGCCCGAAGGCTACGTCCAG
|
|
GFP_control
|
CTACCCCGACCACATGAAGC
|
|
PRIMPOL_WT
|
GATAGCGCTCCAGAGACAAC
|
|
PRIMPOL_del
|
GATAGCGCTCCAGAGAAACA
|
For MFN2 cDNA genotyping, each mutation locus was given a %mutant score calculated as the number of mutant reads divided by total number of reads at that locus. Cells were designated as wildtype if no locus had >50% mutant score, otherwise they were designated as a specific mutant based on which locus had the highest mutant score (ambiguous cells were excluded). Lastly, a flat file was exported containing each picked raft and its assigned genotype.
Using our custom software (FIVTools/ AUC), we joined the modeling and genotyping flat files to find overall accuracy and generate ROC curves for each model. We also generated ‘noise’ ROC curves by shuffling the assigned genotypes. Prediction scores between 0.4 and 0.6 were filtered out (scores near 0.5 meant the specific model was unable to classify these cells). For the data presented in Figure 4e,d, this threshold was further adjusted.
Isogenic Line Production
For clonal cell growth, single live cells were isolated by the Cell Microsystems Air System into 96-well tissue culture plates (TPP 92096), containing 200µl of media per well. As the isogenic lines grew, the entirety of each well was passaged into a plate of larger size (96 to 24 to 12 to 6 well plates from TPP) after reaching ~50% confluency. It took 2 weeks to go from single-cell to 50% confluency in the 96-well plate, and during that time wells were checked for contamination and media level every 2 days. After the cells were plated in the 6-well plate, one third of the cell suspension was taken through DNA extraction for genotyping. One third was frozen down for long term storage, and the remaining third was kept for downstream experiments. The entire process took ~2 months to go from single cells to frozen stocks/genotyping data. Genomic DNA samples were initially genotyped to determine the gRNA(s) present (as described in the preceding sections). Following identification of specific gRNA(s), primers were designed by identifying regions containing gRNA target sites and finding primers that encompassed those regions (Table S5). The genomic DNA samples were then amplified and genotyped a second time using the primer set(s) specific to the target regions in the sample.
Metabolic Analysis
All metabolic analyses were conducted using an Agilent SeahorseXF96 extracellular flux analyzer. Cell culture microplates (Agilent 102601-100) were seeded with 50,000 cells 24 hours prior to running the assay. Sensor cartridges (Agilent 102601-100) were hydrated with sterile water and incubated, along with XF calibrant (Agilent 100840-000), in a non-CO2 incubator 24 hours prior to use. Complete Seahorse assay medium (Agilent 103680-100) was made immediately prior to running the assay according to the manufacturer's instructions. 160µL of XF calibrant was added to the entirety of the plate. The cell culture microplate and sensor cartridge were then incubated at 37°C in a non-CO2 incubator. All assays performed used the Seahorse XF Cell Mito Stress Test Kit (Agilent 103015-100) with Oligomycin 1.5µM, FCCP 1.0µM, Rotenone/Antimycin A 0.5µM, compounds were reconstituted and diluted using complete seahorse medium on the day of the assay. Cell number normalization was performed through image-based counting of cells prior to running the assay (using the InCell as described above).
{kind=link}