To identify somatic variants from uterine corpus endometrial carcinomas (UCEC) and colon adenocarcinomas (COAD), RNA-sequencing alignment files from 587 UCEC and 521 COAD tumor samples were downloaded from The Cancer Genome Atlas (TCGA) and single nucleotide variants were called using the human reference hg38 genome (RVBoost, see methods)12. The median sample had approximately 55,000 called variants, a majority of which are expected to be private, germline variants. We implemented a series of filtering steps aimed at selectively removing germline variants and enrich for somatic variants (Fig. 1A).
The first step was to select for high confidence variants, removing false positives such as sequencing errors, by requiring a read depth of at least 50 reads at the position, with at least 20 reads supporting the alternative allele. This and subsequent filtering steps greatly reduced the number of variants from the original pool (Fig. 1B). The second step was to remove all population variants that have been previously characterized in the dbSNP database, as these variants would highly likely to be germline. The third step was to remove variants that had allelic frequencies between 0.45 and 0.55 (heterozygous) or 0.95 and 1 (homozygous). Tumor samples are often not 100% pure tumor, and additionally, tumors are often mosaic as mutations accumulate in subclones. As a result, the allelic frequencies of somatic variants deviate from germline variants of 0.5 and 1 (Fig. 1C). The number of variants in the resulting list enriched for somatic variants (referred to as the somatic variants for the remainder of the paper) was reduced by a median of 100-fold from the starting variant pool.
The mutational signature of germline variants was determined using the removed variants from filtering steps 2 and 3 (Fig. 1D), and used to estimate the remaining germline population in the final pool of enriched somatic variants. The mutational signature for each sample’s somatic variants was determined in a similar method to the COSMIC signatures and germline signature mentioned above. Linear regression of the trinucleotide frequencies for the somatic variant signatures against those of the germline signature gave a coefficient representing similarity to the germline (see methods). To determine if mutational burden, the number of somatic variants per sequenced Mb, was a function of variability in somatic variant numbers or variability in the filtering process, resulting in left over germline variants, we tried to correlate mutational burden with the similarity to the germline signature (Fig. 1E). Mutational burden was independent of the similarity to the germline signature for all three cancer subtypes, POLɛ, MSI-H, and MSS, suggesting higher mutational burden isn’t due to contaminating germline signatures.
Calling tumor mutational subtype using regression
We next determined if the mutational burden for the POLɛ and MSI-H tumor subtypes, both defined by disruption of DNA integrity mechanisms, was increased relative to MSS tumors, which should only acquire somatic variants from the natural aging process. The mutational burden of POLɛ samples showed significant separation from both MSS and MSI-H tumors (Fig. 2A, p-value < 0.001, unmatched t-test). MSI-H samples also showed statistically significant separation (p-value <0.01, unmatched t-test), however there was substantial overlap between the mutational burden distributions of MSS and MSI-H samples. One concern was that samples sequenced at higher depth would show an increase in TMB, providing a confounding factor to analyzing tumor-only mutational burden. However, comparing the number of megabases with sufficient (>25 read depth required by our variant filtering) for each sample to TMB showed no significant correlation (Fig. 2B, r = 0.015).
Classification of MSS, MSI-H, and POLɛ samples based on supervised approaches
The mutational signatures for MSS, MSI, and POLɛ tumors from COSMIC (Fig. 3A-C), and the germline signature derived from dbSNP variants were compared to tumor-only TCGA samples. TCGA samples with clinical information on MSI and POLɛ status were grouped to make corresponding MSS, MSI-H, and POLɛ mutational signatures from the somatic variants passing through the filtering criteria (Fig. 3D-F). Increased frequency of T to C (green) changes suggests additional germline variants remain after filtering, influencing the signatures of MSS and MSI-H more than POLɛ.
Regression of MSS and MSI-H samples against the germline signature showed no significant difference between the two groups (Fig. 3G). To test if POLɛ and MSI-H samples could be differentiated from MSS cases, we regressed samples’ signatures against MSI-H and POLɛ COSMIC signatures. MSI-H samples had higher coefficients than MSS samples against the MSI-H signature, although the overlap in the coefficient distribution is high (Fig. 3H). POLɛ samples showed a stronger separation of coefficient values from non-POLɛ samples, likely due to the POLɛ signature being less disrupted by contaminating germline variants and distinct from either MSS or MSI-H signatures (Fig. 3I). Next, we tested the performance of the MSI-H and POLɛ signatures derived from the tumor-only TCGA samples against COSMIC signatures. Training and testing sets were randomly selected 10 times and used to calculate the recall and specificity. The POLɛ signature had strong recall and specificity, while MSI-H was moderate, consistant with our previous results (Fig. 4).
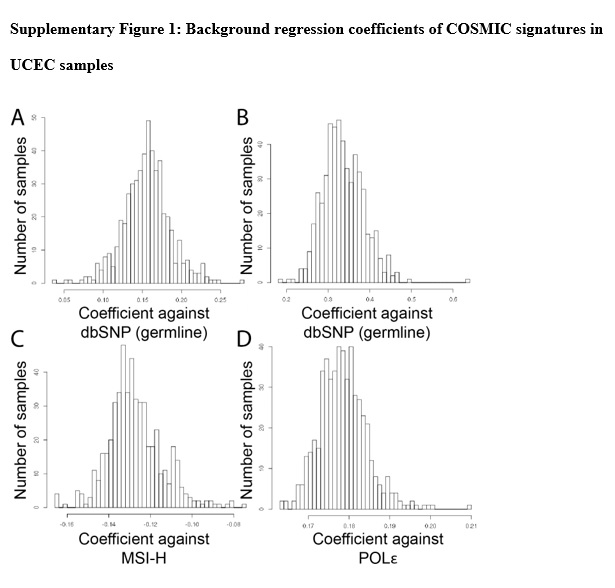
To quantify the per signature bias during regression to RNA samples, we ran 8000 normal GTEx samples through the pipeline. We found that background bias for each signature was relatively distinct and consistent (S. Fig. 1). Germline showed the highest coefficients, consistent with the idea that a majority of the GTEx variants should be germline, familial variants. The distributions of background coefficients can be subsequently used to derive statistics on how far samples are outliers and are likely to be a particular subtype.
Identification of mutational signatures using an unsupervised approach
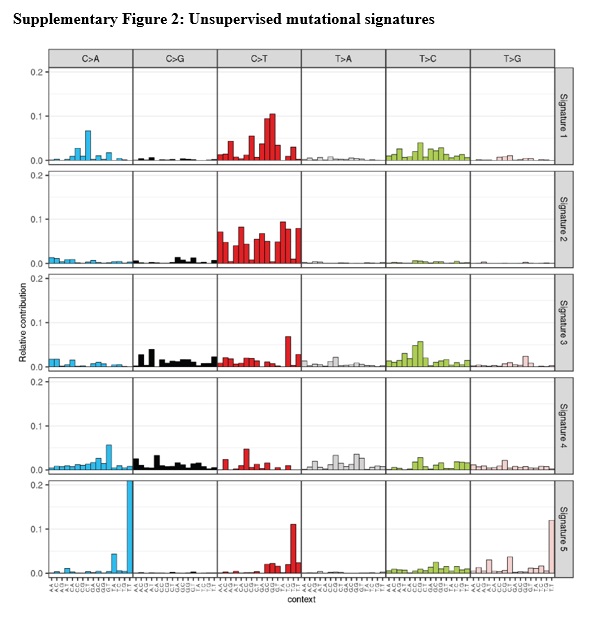
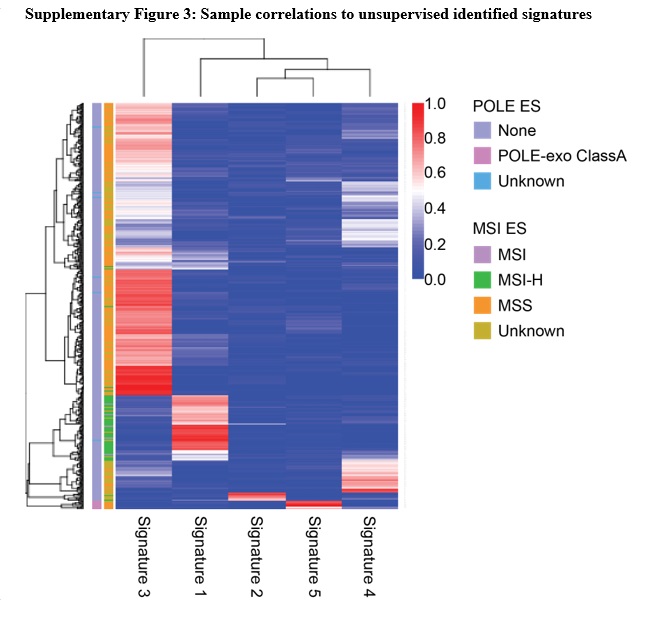
To expand the capabilities of the pipeline to potentially identify rare tumor subtypes we added the mutationalPatterns R package from bioconductor, a previously described, unsupervised approach13. Mutational signatures identified by mutationalPatterns looked comparable to the cosmic signatures used in the supervised approach (S. Fig. 2). Identified signature 3 corresponds to MSS samples, cosmic signature 5; identified signature 1 corresponds to MSI-H, cosmic signature 6; and identified signature 5 corresponds to POLɛ, cosmic signature 10. A majority of samples were explained by the five major signatures identified, and most showing strong correlation to only one signature (S. Fig. 3).
Validation using colorectal TCGA samples
We next wanted to test the performance with signatures generated from the uterine data and applied to a similar dataset of colorectal (CRC) samples from TCGA. The mutational frequencies for each trinucleotide change of MSS, MSI-H, and POLɛ uterine samples were averaged to generate a de-novo signature from the tumor-only, mRNA sequencing data. Generated signatures were then regressed against the mutational frequencies for 521 colorectal tumors, of which 208, had MSS, MSI-H, and POLɛ subtype annotated. The regression coefficients for samples of each subtype were elevated for comparisons against the corresponding de-novo signatures (Fig. 5A-C). The de-novo signatures were able to distinguish between MSS and MSI-H more effectively, and POLɛ just as effectively, compared to regression against the COSMIC signatures (Fig. 5D-F). Taken together, tumor subtype is able to be effectively called when using signatures accounting for the method specific biases encountered from using the RNA of samples lacking a paired normal.
{kind=link}
{kind=link}
{kind=link}
{kind=link}