The algorithm for generating and estimating the probability of possible reaction pathways for multichannel bimolecular interactions was used to predict the reaction products in the reagent ratio of 1:1 and 1:2. Here we have considered the possible reaction pathways of the reaction of amine ((1S,2S,4S)-bicyclo[2.2.1]hept-5-en-2-ylmethanamine (1) with epoxides (2-((cyclohexyloxy)methyl)oxirane (2), 2-(phenoxymethyl)oxirane (3), (N-(oxiran-2-ylmethyl)-N-phenylbenzenesulfonamide 8) in order to explain experimental observed data, which indicate differences in the reactivity of glycidyl ethers and glycidylsulfonamide with framework amines. Based on the proposed algorithm [39], we have investigated the reaction in the reagent ratio of 1:1 and 1: 2. Calculated values of activation barriers indicate a low probability of formation of interaction products of amine (1) with epoxide (8) with a (1:2) reagent ratio due to steric hindrances in the reaction center.
Research Article
Probability of Reaction Pathways of Amine With Epoxides in the Reagent Ratio of 1:1 and 1:2
https://doi.org/10.21203/rs.3.rs-1223033/v1
This work is licensed under a CC BY 4.0 License
You are reading this latest preprint version
multichannel bimolecular interactions
epoxide cycle opening
reagent ratio
glycidyl ethers
Mono- and diepoxide compounds have gained wide popularity due to the broad range of their applications as building blocks for the construction of biologically active systems [1–5], monomers for highly biocompatible and biodegradable polymers [6–11], bases for adhesives [12], and modifiers of polymer compositions [13]. Epoxy cycle opening reactions can be considered as model in the study of complex processes of crosslinking of epoxy monomers with hardeners or their polymerization. In this regard, the study of the mechanisms of opening of the epoxy cycle is undoubtedly an important and urgent task of modern chemistry.
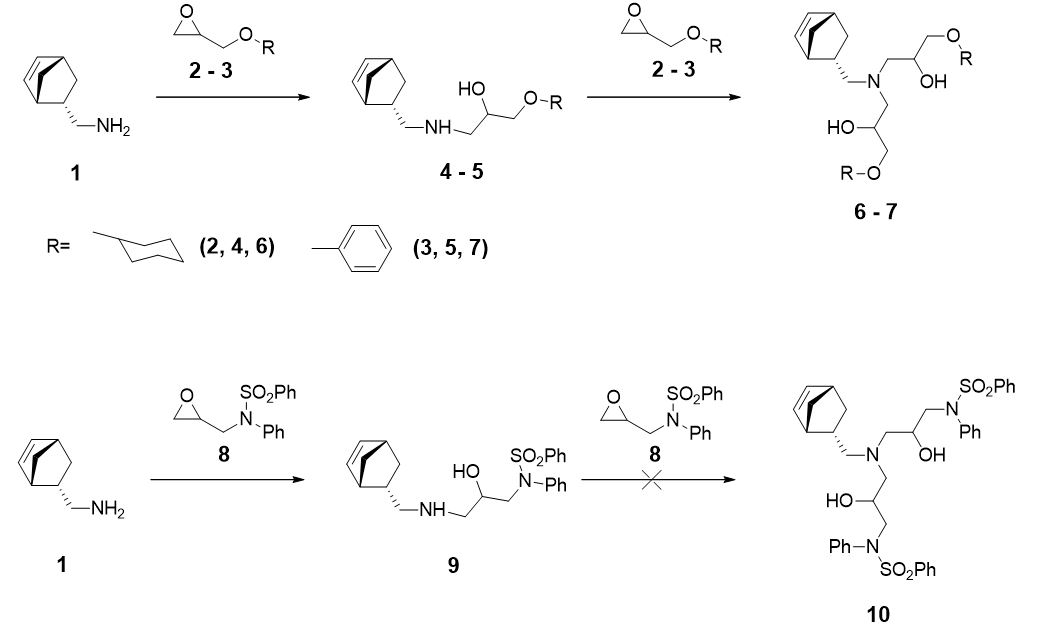
Experimental studies [14, 15] have shown that the interaction of amine ((1S,2S,4S)-bicyclo[2.2.1]hept-5-en-2-ylmethanamine (1) with epoxides (2-((cyclohexyloxy)methyl)oxirane (2), 2-(phenoxymethyl)oxirane (3) forms a mixture of products that correspond to reactions with a ratio of reagents 1:1 and 1:2 (amino alcohols 4, 5 and 6, 7, respectively, Scheme 1). While in the case of epoxide (N-(oxiran-2-ylmethyl)-N-phenylbenzenesulfonamide 8) the reaction ends with the formation of product (9), that corresponds to a reagent ratio 1:1.
For such compounds, the study of mechanism and the dependence of the ratio of products on the ratio of the initial reagents was already published [15]. The aim of this work was to study the possible reaction pathways of the aminolysis of epoxides (2, 3, 8) considering the conformational lability of the reagents in order to explain the experimentally observed formation of products (6, 7) and the absence of product (10).
Over the last decades several computer assisted automatic methods for systematic searching reaction pathway and determining reaction mechanisms have been proposed. Among these methods the following should be noted: anharmonic downward distortion following (ADDF) method [16–19]; artificial force-induced reaction (AFIR) method [20–24]; transition state search using chemical dynamics simulations (TSSCDS) method [25–28]; the ZStruct2 method developed by Zimmerman’s research group [29–31]; and molecular dynamics and coordinate driving (MD/CD) method proposed by Li and coworkers [32]. Since each method for searching reaction pathway aims to provide a balance between high accuracy, comprehensive potential energy surface search and low computational costs, the development of new, efficient algorithms for studying reaction paths remains an urgent issue for computational chemistry.
The program PCModel v 9.2 [33] was used to perform conformational analysis at the MMX force field level [34]. Conformational search calculations were carried out using the GMMX technique.
Semi-empirical calculations by РМ7 [35] method were carried out using MOPAC2016 [36] program, DFT calculations at the M062X/6-31G(d) [37] level of theory were performed using the Gaussian 16 program [38]. Barrier heights evaluated at semi-empirical level were calculated using PM7-TS method as single point calculations based on PM7 geometry. Vibrational frequencies were calculated for all stationary points to confirm whether the optimized geometry corresponds to a minimum or a TS. The solvent effect, where mentioned, was taken into account by consideration of a solute molecule surrounded by several solvent molecules.
In order to study the features of aminolysis of epoxides (2, 3, 8) we applied the algorithm for generation and assessment of probability of possible reaction pathways for multiple channel bimolecular interactions proposed in our previous study [39], which was devoted to the interaction of epoxide (2) with amine (1) to obtain glycidyl ether (4).
This algorithm includes conformational search for reaction intermediate using Molecular Mechanics (MMX) approach, based on the obtained conformation construction of structures of transition states and pre-reaction complexes, and calculation of activation energies to further determine the probable reaction pathways.
In this work, initially, we studied the reaction of amine (1) with epoxides (3, 8) in the ratio of reactants 1:1. Then the investigation of the reaction in the ratio of reagents 1:2 was carried out according to the analogous procedure.
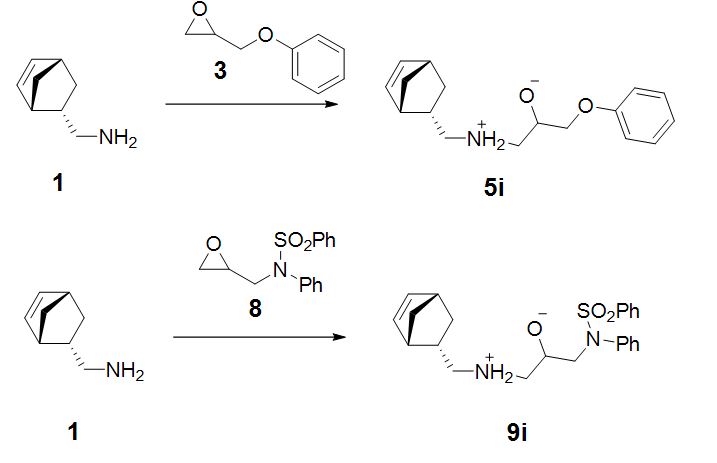
The reaction we have considered proceeds through SN2-like mechanism forming bipolar ion (5i,9i) as an intermediate on the rate-limiting endorgonic stage (Scheme 2).
Located conformations of bipolar ions (5i, 9i) could be straightforward applied for construction of starting geometry for optimization of transition states of the corresponding reactions. This statement follows from Hammond’s postulate which states that in the case of endorgonic stage the geometry of the intermediate should be close to the geometry of the transition state.
The proposed strategy consists of the following steps:
(I) Conformational analysis of intermediates (5i, 9i) using Molecular Mechanics (MMX) approach.
(II) Construction of starting geometries for TS localization based on the most stable conformations of the intermediates (5i, 9i) by setting up C-O and C-N bond lengths close to model aminolysis reaction [39].
(III) Locating TSs and pre-reaction complexes at semi-empirical (PM7) level followed by calculations of activation barriers using PM7-TS method. In the case when the optimization of TSs yielded the structures already existing in the set, they were excluded from the sample.
(IV) Locating TSs, pre-reaction complexes at M062X/6-31G(d) level of theory. As starting geometries, we have used TS geometries that had been obtained in the step (III). Similarly, to step (III), the repeating structures were excluded from the sample.
(V) The reaction paths obtained by M062X/6-31G(d), whose contributions to the overall rate constant were the largest, have been selected for calculation of reaction paths considering the influence of the solvent.
Let us now apply the strategy described in the steps I-V in more details for reaction of amine (1) with epoxides (3, 8). The assumed ratio of reactants is 1:1.
Conformational analysis
Calculations were carried out using MMX force field within GMMX technique as implemented in PCModel v 9.2 program.
Since the conformational transitions in the intermediates (5i,9i) are associated with rotation around the bonds, in order to generate the initial structures by the GMMX method the algorithm that randomly selects a subset of the bonds intended for rotation was chosen. The N‑C‑C‑O torsion angle value was fixed at 180° because this angle value corresponds to the trans-opening state of the epoxy ring. All other torsion angles were used to create conformations by the GMMX method. Based on the previous study [39] for GMMX calculation the value of energy window equal to 40 kJ/mol was chosen.
Of all the found conformations we were interested in the most stable conformer with the highest population. The total number of conformations selected for further localization of transition states for possible pathways was equal to 15 in case of intermediate (5i) and 11 in case of intermediate (9i).
Locating of TSs and pre-reaction complexes at semi-empirical (PM7) level
Geometry of conformations from previous step has been modified by setting up length of forming N‑C and breaking O‑C bonds lengths equal to 1.782 Å and 1.999 Å, respectively. This corresponds to the transition state geometry parameters of model reaction (interaction of methylamine with oxirane) (see [39]).
After localization of TS structures and exclusion of repeating ones the pre-reaction complexes and energy barriers were calculated. Table 1 shows obtained activation barrier values for the first step of aminolysis reaction and contributions of each routes to the total reaction rate constant. The overall contributions to the total reaction rate constant (ki) were calculated using equation (1).

According to PM7 method the largest contribution of 66.9% to the total reaction rate is made by TS that corresponds to the reaction path number 4 in the case of intermediate (5i). For intermediate (9i) the largest contributions to the total reaction rate equal to 45.0 and 43.0 % are made by TSs that correspond to the reaction paths 6 and 7.
For all these transition states, the presence of a hydrogen bond (NH•••O) is observed, which leads to the stabilization of the structure (Figure 1).
Table 1. The values of PM7 heat of formation (kJ/mol), the activation energy (kJ/mol) calculated at PM7-TS and М062Х/6-31G(d) levels of theory. Conformers of intermediate (5i, 9i) for possible reaction channels and the contribution of located pathways to overall rate constant of the reaction of amine (1) with epoxide (3, 8) are included.
|
Intermediate |
Pathway |
PM7 |
М062Х/6-31G(d) |
||
|
∆H≠ PM7-TS, kJ/mol |
ki,% |
∆E≠zpc, kJ/mol |
ki,% |
||
|
5i |
1 |
156.4 |
0.0 |
149.3 |
0.8 |
|
2 |
139.2 |
9.3 |
- |
- |
|
|
3 |
149.1 |
0.2 |
- |
- |
|
|
4 |
134.3 |
66.9 |
142.3 |
14.4 |
|
|
5 |
148.8 |
0.2 |
141.9 |
16.9 |
|
|
6 |
151.0 |
0.1 |
151.4 |
0.4 |
|
|
7 |
140.7 |
5.0 |
149.5 |
0.8 |
|
|
8 |
137.5 |
18.0 |
138.5 |
66.7 |
|
|
9 |
147.4 |
0.3 |
145.2 |
4.4 |
|
|
9i |
1 |
136.9 |
3.7 |
147.0 |
0.0 |
|
2 |
135.0 |
8.0 |
129.2 |
19.2 |
|
|
3 |
159.0 |
0.0 |
136.0 |
1.3 |
|
|
4 |
149.1 |
0.0 |
131.7 |
7.3 |
|
|
5 |
143.7 |
0.2 |
141.4 |
0.1 |
|
|
6 |
130.7 |
45.0 |
137.2 |
0.8 |
|
|
7 |
130.8 |
43.1 |
127.8 |
33.7 |
|
|
8 |
157.0 |
0.0 |
127.6 |
37.6 |
|
|
9 |
191.4 |
0.0 |
147.0 |
0.0 |
|
Study of reaction in vacuo at M062X/6-31G(d) level of theory
The starting geometries for the localization of transition states were structures obtained by the PM7 method.
All of 18 possible reaction paths obtained for intermediates (5i, 9i) by the PM7 method were studied at the M062X/6-31G(d) level of theory. After excluding TSs with the same geometries, 7 TS conformations were obtained for intermediate (5i) and 9 for intermediate (9i).
Starting geometry for optimization of pre-reaction complexes was generated by displacement of atoms in the TS structures along imaginary normal vibrational mode. The activation barriers were calculated for each reaction channel (Table 1).
The largest contribution of 66.7% to the total reaction rate is made by TS of intermediate (5i) that corresponds to path 8. In accordance with the results for intermediate (9i) the largest contributions to the reaction rate (33.7 and 37.6%) are from the TS conformations that correspond to paths 7 and 8 respectively. The structures of these TS conformations are shown in Figure 2.
The most energetically favorable reaction paths were also examined considering solvent effects.
Study of reaction with 1:2 reagent ratio in vacuo
The procedure for studying the aminolysis reaction with 1:2 reagent ratio was similar to that described above for 1:1 reagent ratio. Let us discuss the obtained results.
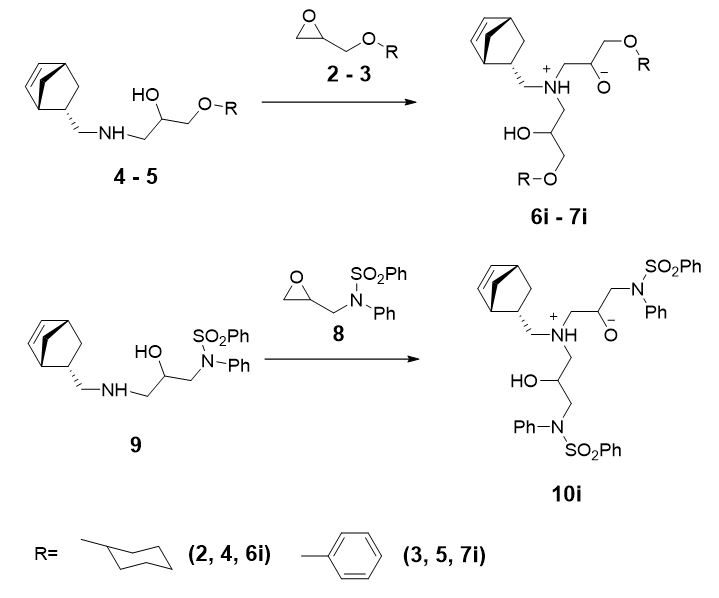
At the first step we carried out conformation search for intermediates (6i,7i,10i) (Scheme 3). For these intermediate 6i, 7i, 10i the number of unique conformations was 41, 7, and 8, respectively.
At the second step of the study, the transition states and pre-reaction complexes for each found conformer of all three intermediates were localized at PM7 and М062Х/6-31G(d) levels of theory. Subsequently, the activation barriers of possible reaction channels and the contribution of each TS to the total reaction rate were calculated. As can be seen from Table 2 the results of the semi-empirical and the DFT methods are slightly different. The largest contribution to the overall reaction rate for intermediate 6i is from the TSs number 22 (80.4%) obtained by PM7 method. However, at the М062Х/6-31G(d) level the TS 24 has the largest input of 21.8%. For intermediate 7i at the PM7 level the largest contribution to the overall reaction rate (94.9%) is from the TS number 4. Interestingly, М062Х/6-31G(d) method predicts TS number 1 to have the highest contribution. In case of intermediate 10i the largest contributions to the overall reaction rate equal to 25.7 and 54.3% are from TSs number 6 and 7 as obtained by PM7 method. М062Х/6-31G(d) method showed that TSs number 1 and 7 have the greatest contribution to the rate of reaction equal to 59.1 and 40.0%, respectively. The structures of these TS conformations are shown in Figure 3.
Study of reaction with explicit consideration of solvent at M062X/6-31G(d) level of theory
Since the experimental reaction was carried out in the presence of the protic solvent 2-propanol [15], the specific solvation with solvent was modelled in this study by adding two molecules of alcohol to the investigated molecular system. The initial position of solvent molecules in the transition state can be characterized by the formation of hydrogen bonds between alcohol and epoxide, alcohol and alcohol, alcohol and amine molecules. The structures of the transition states of the formation of aminoalcohols in the presence of the solvent (5,6,7,9,10) are given in Figure 4, and their cartesian coordinates are included in the Supporting Information.
The participation of two alcohol molecules leads to the relay transfer of a proton from the nitrogen atom of amine to oxygen atom of epoxide along the chain of solvent molecule. Thus, simultaneous activation of epoxide and amine by two solvent molecules occurs and leads to lower activation barrier of the reaction by 23.2 – 64.8 kJ/mol (Table 3).
All reactions with a reagent ratio of 1:1 are characterized by barriers from 127.6 to 139.0 kJ/mol in vacuo, and from 66.6 to 91.5 kJ/mol within explicit consideration of solvent molecules. For reactions with a reagent ratio of 1:2 the values of ΔE≠ are also close to those predicted for the 1:1 ratio and range from 117.5 to 128.5 kJ/mol in vacuo and 78.1 to 94.3 kJ/mol within explicit consideration of solvent molecules.
In the case of the interaction of epoxide (8) with amine (1), the value of the activation energy at the reagent ratio 1:2 is higher by 11.6 – 27.7 kJ/mol than the corresponding values predicted for the ratio of 1:1. The obtained results are consistent with the experimental data, according to which in this case only the product of the interaction with the 1:1 reagent ratio is formed. The plausible reason for the observed increase in the energy barrier is the large steric hindrance for the interaction of bulk reagents in a process characterized by 1:2 reagent ratio. The obtained activation barriers are consistent with experimental data, which indicate differences in the reactivity of glycidyl ethers and glycidylsulfonamide with framework amines.
Table 2. The values of PM7 heat of formation (kJ/mol), the activation energy (kJ/mol) calculated at PM7-TS and М062Х/6-31G(d) levels of theory. Conformers of intermediate (6i, 7i, 10i) for possible reaction channels and the contribution of located pathways to overall rate constant of the reactions of glycidyl ethers (4,5) with epoxide (2,3) and glycidylsulfonamide (9) with epoxide (8) are included.
|
Intermediate |
Pathway |
PM7 |
М062Х/6-31G(d) |
||
|
∆H≠ PM7-TS, kJ/mol |
ki,% |
∆E≠zpc, kJ/mol |
ki,% |
||
|
6i |
1 |
160.2 |
0.0 |
131.9 |
5.6 |
|
2 |
146.2 |
9.9 |
132.8 |
4.0 |
|
|
3 |
187.5 |
0.0 |
139.7 |
0.2 |
|
|
4 |
181.2 |
0.0 |
143.7 |
0.1 |
|
|
5 |
150.1 |
2.1 |
150.7 |
0.0 |
|
|
6 |
173.4 |
0.0 |
147.1 |
0.0 |
|
|
7 |
152.1 |
0.9 |
132.6 |
4.2 |
|
|
8 |
149.8 |
2.4 |
131.9 |
5.5 |
|
|
9 |
160.4 |
0.0 |
129.7 |
13.5 |
|
|
10 |
159.1 |
0.1 |
132.6 |
4.2 |
|
|
11 |
160.8 |
0.0 |
143.1 |
0.1 |
|
|
12 |
154.4 |
0.4 |
132.8 |
3.9 |
|
|
13 |
172.1 |
0.0 |
138.3 |
0.4 |
|
|
14 |
162.3 |
0.0 |
130.7 |
9.1 |
|
|
15 |
176.3 |
0.0 |
141.5 |
0.1 |
|
|
16 |
165.8 |
0.0 |
142.3 |
0.1 |
|
|
17 |
164.8 |
0.0 |
133.8 |
2.6 |
|
|
18 |
185.1 |
0.0 |
148.5 |
0.0 |
|
|
19 |
166.9 |
0.0 |
143.1 |
0.1 |
|
|
20 |
177.4 |
0.0 |
172.8 |
0.0 |
|
|
21 |
201.0 |
0.0 |
146.6 |
0.0 |
|
|
22 |
141.0 |
80.4 |
132.9 |
3.8 |
|
|
23 |
155.0 |
0.3 |
145.5 |
0.0 |
|
|
24 |
167.7 |
0.0 |
128.5 |
21.8 |
|
|
25 |
199.3 |
0.0 |
141.7 |
0.1 |
|
|
26 |
190.7 |
0.0 |
164.1 |
0.0 |
|
|
27 |
163.3 |
0.0 |
142.6 |
0.1 |
|
|
28 |
161.3 |
0.0 |
148.9 |
0.0 |
|
|
29 |
160.3 |
0.0 |
132.5 |
4.4 |
|
|
30 |
149.2 |
2.9 |
148.5 |
0.0 |
|
|
31 |
210.5 |
0.0 |
132.1 |
5.2 |
|
|
32 |
162.7 |
0.0 |
139.4 |
0.3 |
|
|
33 |
211.2 |
0.0 |
146.2 |
0.0 |
|
|
34 |
182.3 |
0.0 |
139.8 |
0.2 |
|
|
35 |
165.3 |
0.0 |
156.9 |
0.0 |
|
|
36 |
161.3 |
0.0 |
138.2 |
0.4 |
|
|
37 |
155.1 |
0.3 |
130.5 |
9.9 |
|
|
38 |
155.5 |
0.2 |
146.3 |
0.0 |
|
|
39 |
224.8 |
0.0 |
149.9 |
0.0 |
|
|
40 |
160.1 |
0.0 |
155.0 |
0.0 |
|
|
41 |
245.0 |
0.0 |
158.7 |
0.0 |
|
|
7i |
1 |
163.6 |
3.3 |
123.4 |
99.5 |
|
2 |
207.7 |
0.0 |
139.8 |
0.1 |
|
|
3 |
185.9 |
0.0 |
139.5 |
0.2 |
|
|
4 |
155.3 |
94.9 |
143.6 |
0.0 |
|
|
5 |
165.2 |
1.7 |
148.9 |
0.0 |
|
|
6 |
183.9 |
0.0 |
138.6 |
0.2 |
|
|
7 |
242.0 |
0.0 |
162.3 |
0.0 |
|
|
10i |
1 |
233.1 |
0.0 |
117.5 |
59.1 |
|
2 |
179.1 |
0.0 |
155.4 |
0.0 |
|
|
3 |
153.3 |
16.8 |
134.1 |
0.1 |
|
|
4 |
157.5 |
3.1 |
128.2 |
0.8 |
|
|
5 |
235.0 |
0.0 |
137.2 |
0.0 |
|
|
6 |
152.3 |
25.7 |
151.7 |
0.0 |
|
|
7 |
150.4 |
54.3 |
118.5 |
40.0 |
|
|
8 |
200.5 |
0.0 |
142.6 |
0.0 |
|
Table 3. Сalculated values of activation energy for the most favorable pathways of aminolysis reactions with a reagent ratio of 1:1 and 1:2 calculated by M062X/6-31G(d)
|
Structure of epoxides |
Intermediate |
Pathway |
reagent ratio |
in vacuo for rate-limiting stage |
Explicit consideration of two 2-propanol molecules |
|
ΔE≠kJ/mol |
ΔE≠kJ/mol |
||||
|
2 |
- |
- |
1:1 |
139.0* |
74.2 |
|
6i |
24 |
1:2 |
128.5 |
80.1 |
|
|
3 |
5i |
8 |
1:1 |
138.5 |
84.0 |
|
7i |
1 |
1:2 |
123.4 |
72.0 |
|
|
8 |
9i |
8 |
1:1 |
127.6 |
66.6 |
|
7 |
1:1 |
127.8 |
91.5 |
||
|
10i |
1 |
1:2 |
117.5 |
94.3 |
|
|
7 |
1:2 |
118.5 |
78.1 |
*Data from [39]
Conformational analysis of primary products of reactions could be considered as general procedure for generation of transition state structures in the case of multiple channel reactions. Method of generation of transition state structures for multiple channel reactions that considers the high conformational mobility of the reactants and the possibility of varying their orientation during the reaction was tested in this study. Conformational analysis of investigated epoxy esters derivatives, conducted using method of molecular mechanics MMX, revealed structures with the lowest energies – the most stable conformers of primary products of epoxy compounds aminolysis. Transition state structures for the reaction of aminolysis of glycidyl ethers and glycidylsulfonamides with frame amines were localized using quantum-chemical methods PM7 and M062X/6-31G(d). Formation of 1:1 and 1:2 products of glycidyl eters aminolysis are determines by steric effect and formation of N-H∙∙∙O hydrogen bond in transition state. Calculated values of activation barriers indicate a low probability of formation of interaction products of amine with epoxyglycidylsulfonamides with a 1:2 reagent ratio, which is consistent with and explains experimental data.
Funding
The computation time was provided by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562 and XSEDE award allocation Number TG-DMR110088. This study was supported by the Ministry of Education and Science of Ukraine (grant 0119U100724).
Conflicts of interest/Competing interests
The authors declare that they have no conflict of interest.
Availability of data and material
Additional data (Cartesian Coordinates and Gibbs free energies of TS conformations from the M062X/6-31G(d) calculation) are available as Supplementary Information.
Code availability
Not applicable.
Authors’ contributions
All authors contributed to the study conception and design. Material preparation, data collection, computer simulation were performed by Iryna O. Borysenko. Analysis of the obtained data was performed by Iryna O. Borysenko, Sergiy I. Okovytyy and Jerzy Leszczynski. The first draft of the manuscript was written by Iryna O. Borysenko and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
- Bergmeier S (2000) The Synthesis of Vicinal Amino Alcohols. Tetrahedron 56(17): 2561–2576
- Cristau H-J, Pirat J-L, Drag M, Kafarski P (2000) Regio- and stereoselective synthesis of 2-amino-1-hydroxy-2-aryl ethylphosphonic esters.Tetrahedron Lett. 41: 9781–9785
- Kasyan L, Okovytyy S, Kasyan A (2004) Reactions of Alicyclic Epoxy Compounds with Nitrogen-Containing Nucleophiles Russ. J Org Chem 40: 1–34
- Kasyan L, Palchikov V (2010) Cage-like amino alcohols. synthesis, reactions, and application. Russ. J Org Chem 46: 1–42
- Sayer J, Chadha A, Agarwal S, Yeh H, Yagi H, Jerina D (1991) Covalent nucleoside adducts of benzo[a]pyrene 7,8-diol 9,10-epoxides: structural reinvestigation and characterization of a novel adenosine adduct on the ribose moiety. J Org Chem 56: 20–29
- Scharfenberg M, Hilf J, Frey H (2018) Functional Polycarbonates from Carbon Dioxide and Tailored Epoxide Monomers: Degradable Materials and Their Application Potential. Advanced Functional Materials 28(10): 1704302. doi:10.1002/adfm.201704302
- Wang R, Schuman T (2013) Vegetable oil-derived epoxy monomers and polymer blends: A comparative study with review. Express Polymer Letters 7(3): 272–292. doi:10.3144/expresspolymlett.2013.25
- Ma S, Liu X, Fan L, Jiang Y, Cao L, Tang Z, Zhu J (2013) Synthesis and Properties of a Bio-Based Epoxy Resin with High Epoxy Value and Low Viscosity. ChemSusChem 7(2): 555–562. doi:10.1002/cssc.201300749
- Mangold C, Obermeier B, Wurm F, Frey H (2011) From an Epoxide Monomer Toolkit to Functional PEG Copolymers With Adjustable LCST Behavior. Macromolecular Rapid Communications 32(23): 1930–1934. doi:10.1002/marc.201100489
- Huang K, Liu Z, Zhang J, Li S, Li M, Xia J, Zhou Y (2014) Epoxy Monomers Derived from Tung Oil Fatty Acids and Its Regulable Thermosets Cured in Two Synergistic Ways. Biomacromolecules 15(3): 837–843. doi:10.1021/bm4018929
- Ng F, Couture G, Philippe C, Boutevin B, Caillol S (2017) Bio-Based Aromatic Epoxy Monomers for Thermoset Materials. Molecules 22(1): 149. doi:10.3390/molecules22010149
- Stadler B, Tin S, Kux A, Grauke R, Koy C, Tiemersma-Wegman T, de Vries J (2020) Co-oligomers of renewable and “inert” 2-MeTHF and propylene oxide for use in bio-based adhesives. ACS Sustainable Chemistry & Engineering 8(35): 13467–13480. doi:10.1021/acssuschemeng.0c04450
- Cerit A, Marti M, Soydal U, Kocaman S, Ahmetli G (2016) Effect of Modification with Various Epoxide Compounds on Mechanical, Thermal, and Coating Properties of Epoxy Resin. International Journal of Polymer Science 7: 1–13. doi:10.1155/2016/4968365
- Kasyan L, Pridma S, Palchikov V, Karat L, Turov A, Isayev O (2010) Reaction of bicyclo[2.2.1]hept-5-ene-endo-2-ylmethylamine and nitrophenyl glycidyl ethers. Journal of Physical Organic Chemistry 24(8): 705–713. doi:10.1002/poc.1815
- Palchikov V, Svyatenko L, Plakhotnii I, Kas’yan L (2013) Experimental and Theoretical Study of the reaction between bicyclo[2.2.1]hept-5-en-endo-2-ylmethylamine and 2-[(2-allylphenoxy)methyl]oxirane. Zhurnal Organicheskoi Khimii 49:704−708
- Ohno K, Maeda S (2004) A scaled hypersphere search method for the topography of reaction pathways on the potential energy surface. Chem Phys Lett 384:277–282
- Maeda S, Ohno K (2005) Global mapping of equilibrium and transition structures on potential energy surfaces by the scaled hypersphere search method: applications to ab initio surfaces of formaldehyde and propyne molecules. J Phys Chem A 109:5742–5753
- Ohno K, Maeda S (2006) Global reaction route mapping on potential energy surfaces of formaldehyde, formic acid, and their metal-substituted analogues. J Phys Chem A 110:8933–8941
- Maeda S, Ohno K, Morokuma K (2013) Systematic exploration of the mechanism of chemical reactions: the global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys Chem 15:3683–3701
- Maeda S, Morokuma K (2010) Communications: a systematic method for locating transition structures of A + B = X type reactions. J Chem Phys 132:241102
- Maeda S, Morokuma K (2011) Finding reaction pathways of type A + B = X: toward systematic prediction of reaction mechanisms. J Chem Theory Comput 7:2335–2345
- Maeda S, Abe E, Hatanaka M, Taketsugu T, Morokuma K (2012) Exploring Potential Energy Surfaces of Large Systems with Artificial Force Induced Reaction Method in Combination with ONIOM and Microiteration. J Chem Theory Comput 8:5058−5063
- Maeda S, Harabuchi Y, Takagi M, Taketsugu T, Morokuma, K (2016) Artificial force induced reaction (AFIR) method for exploring quantum chemical potential energy surfaces. Chem Rec 16:2232–2248
- Maeda S, Harabuchi Y, Takagi M, Saita K, Suzuki K, Ichino T, Sumiya Y, Sugiyama K, Ono Y (2017) Implementation and performance of the artificial force induced reaction method in the GRRM17 program. J Comput Chem 39:233–250
- Martínez-Núñez E (2015) An automated transition state search using classical trajectories initialized at multiple minima. Phys Chem 17:14912–14921
- Martínez-Núñez E (2015) An automated method to find transition states using chemical dynamics simulations. J Comput Chem 36:222–234
- Ferro-Costas D, Martínez-Núñez E, Rodríguez-Otero J, Cabaleiro-Lago E, Estévez CM, Fernández B, Fernández-Ramos A, Vázquez SA (2018) Influence of Multiple Conformations and Paths on Rate Constants and Product Branching Ratios. Thermal Decomposition of 1-Propanol Radicals. J Phys Chem A 122:4790–4800
- Varela JA, Vazquez SA, Martinez-Nunez E (2017) An automated method to find reaction mechanisms and solve the kinetics in organometallic catalysis. Chem Sci 8:3843–3851
- Dewyer AL, Zimmerman PM (2017) Finding reaction mechanisms, intuitive or otherwise. Org Biomol Chem 15:501–504
- Ludwig JR, Phan S, McAtee CC, Zimmerman PM, Devery JJ, Schindler CS (2017) Mechanistic investigations of the iron(III)-catalyzed carbonyl-olefin metathesis. J Am Chem Soc 139:10832–10842
- Dewyer AL, Zimmerman PM (2017) Simulated mechanism for palladium-catalyzed, directed γ-arylation of piperidine. ACS Catal 7:5466–5477
- Yang M, Zou J, Wang G, Li S (2017) Automatic Reaction Pathway Search via Combined Molecular Dynamics and Coordinate Driving Method. J Phys Chem A 121:1351−1361
- PCModel V 9.0. Molecular Modeling Software for Windows Operating System Apple Macintosh OS Linux and Unix. 2004. Serena Software Box 3076 Bloomington, IN 47402-3076 (812)-333-0823.
- Gajewski JJ, Gilbert KE, McKelvey J (1990) MMX: an enhanced version of MM2, in Advances in Molecular Modeling. ed. D. Liotta, JAI Press, Greenwich, CT 2:65
- Stewart JJP (2013) Optimization of parameters for semi-empirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J Molec Modeling 19:1-32
- MOPAC2016, James J. P. Stewart, Stewart Computational Chemistry, Colorado Springs, CO, USA, HTTP://OpenMOPAC.net (2016)
- Zhao Y, Truhlar DG (2006) Comparative DFT study of van der Waals complexes: Rare-gas dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers. J Phys Chem 110:5121-5129
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JAJr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT
- Borysenko I, Sviatenko L, Okovytyy S, Leszczynski J (2021) Efficient approach for exploring the multiple-channel bimolecular interactions of conformationally flexible reagents. Epoxide ring opening reaction. Structural Chemistry 32: 581–589 https://doi.org/10.1007/s11224-020-01663-0
Please see the Supplementary Files for the Scheme 1, 2 and 3.
{kind=link}
{kind=link}
{kind=link}