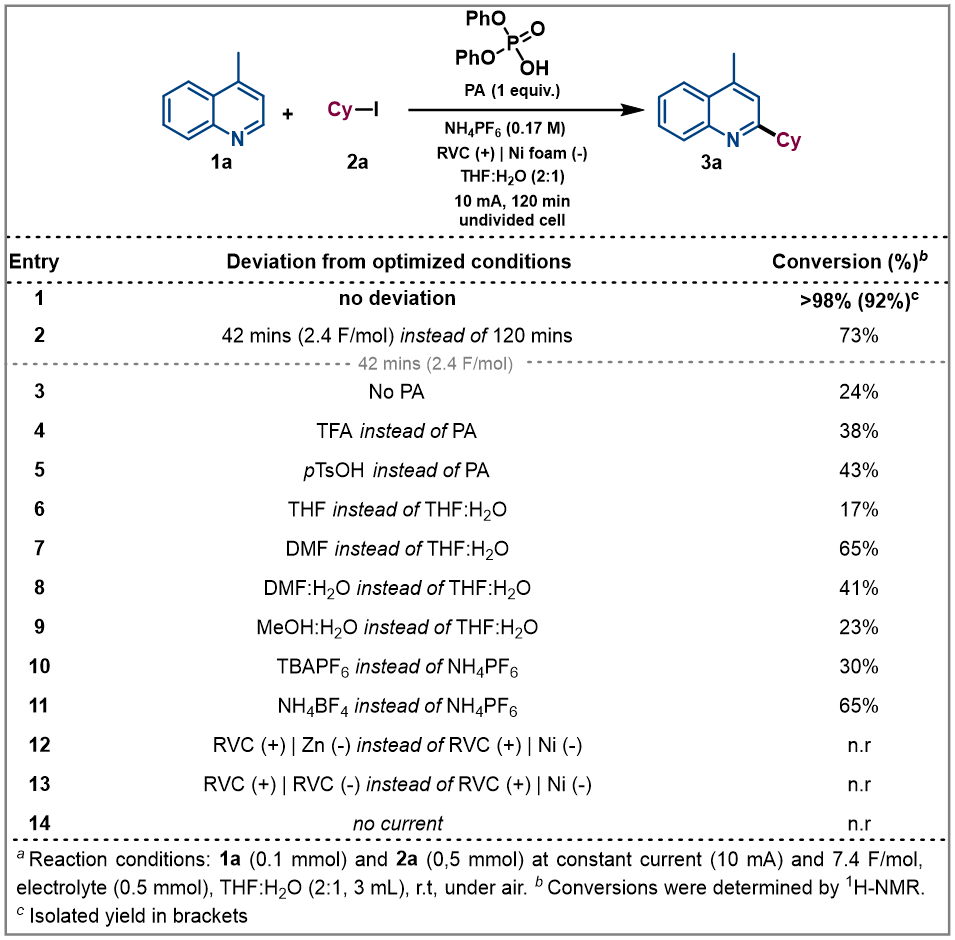
Optimization of the model reaction. We began our investigations by studying the reaction of 4-methylquinoline (1a) as model substrate with cyclohexyl iodide (2a) as radical precursor (Table 1). By using diphenyl phosphate (PA), NH4PF6 as electrolyte under 10 mA constant current for 120 mins in an undivided cell, the alkylated quinoline derivative 3a was isolated in excellent yield (92%, entry 1). Operationally, the setup uses a simple commercial potentiostat, without any of the ordinary precautions required in radical chemistry, such as meticulous exclusion of O2 or water, since the reaction takes place in a mixture of THF: H2O under air. For the development of a synthetically useful electrochemical Minisci-type alkylation, various parameters such as the additive or promoter, solvent, electrolyte, electrode material and electrochemical parameters have been studied and summarized in Table 1 (see Supplementary Information for details). When the reaction was stopped after 42 mins (2.4 F/mol), 73% conversion of the desired alkylated heteroarene was obtained (entry 2). In presence of other acids such as trifluoroacetic acid (TFA) or para-toluenesulfonic acid (pTsOH) the reaction took place less efficiently and lower reactivities were observed (entries 4 and 5), which demonstrates the special capability of the PA to carry out the activation of quinoline derivatives.17 Different solvent systems commonly used under electrochemical reaction conditions (entries 6-9, see Supplementary Information for details) were then evaluated, showing that H2O as co-solvent was necessary to competently perform the alkylation. Modification of the electrolyte showed that the use of a Brønsted acid-based electrolyte had a considerable effect on the reaction as it might be also involved in the activation of the heterocycle (entries 10 and 11). The electrode material selection had a great impact on the reaction as well. Thus, Zn or carbon-based electrodes (RVC) cathodes did not promote the reaction (entries 12 and 13). Finally, the reaction did not take place in absence of electrical current (entry 14).
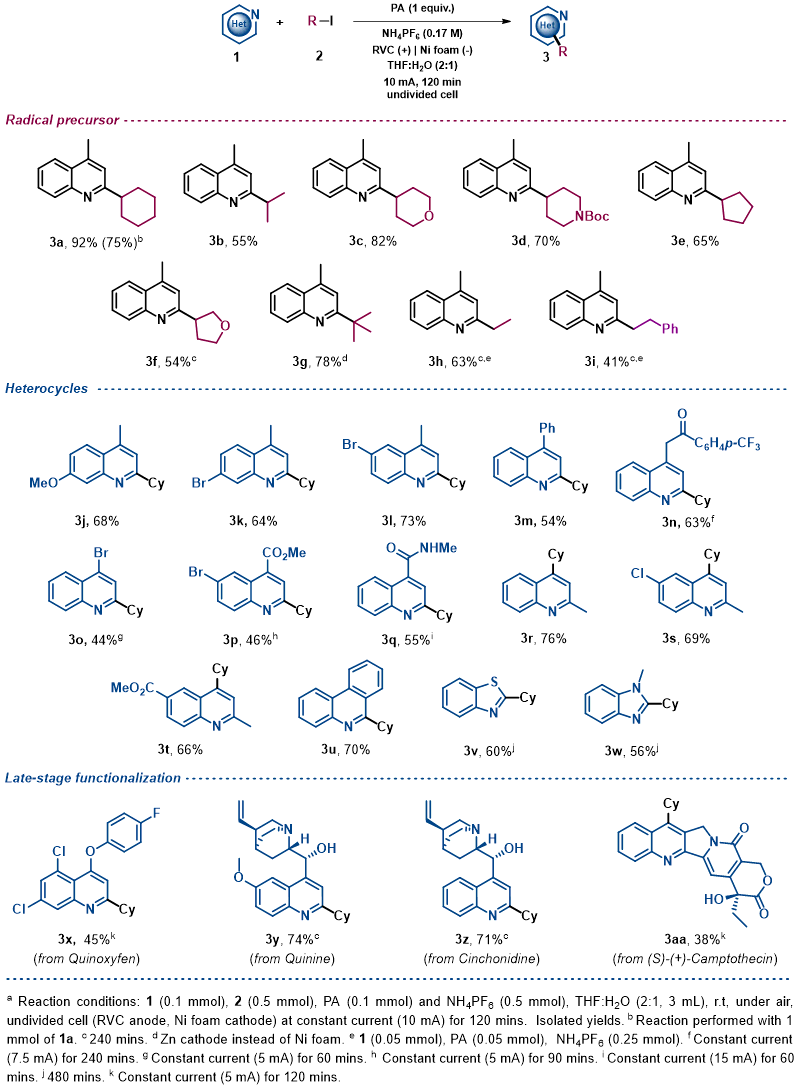
Substrate Scope. Once the reaction conditions had been optimized, a variety of alkyl iodides were tested under the electrochemical reaction conditions in an undivided cell with a readily available RVC anode and a nickel foam cathode. The initial aim of this study was to evaluate the generality of the system using 4-methylquinoline as model substrate (Table 2). This exploration demonstrated that secondary alkyl radical precursors efficiently performed the alkylation in good yields (3a-3f). Notably, secondary alkyl iodides bearing heteroatoms were very well tolerated (3c, 3d and 3f). A sterically encumbered tertiary alkyl radical also led to the desired alkylated quinoline with very good yield (3g). Furthermore, primary alkyl radicals were competently generated under the electrochemical conditions presented and furnished the desired 2-substituted quinolines in good yields (3h and 3i). We then explored the heteroaromatic radical acceptor. Firstly, differently substituted quinolines were subjected to the optimized reaction conditions. Thus, methoxy- and bromide-bearing 4-methyl-substituted quinolines provided the desired alkylated heterocyclic systems (3j, 3k and 3l). Notably, aromatic bromides were perfectly compatible with the electrochemical reaction conditions, which should be highlighted as the presence of bromides are very often circumvented in the substrate scope evaluation of previous Minisci-type alkylation studies using alkyl halides,[28, 30–33] probably to avoid other reduction byproducts. We then evaluated the substitution at the 4-position of the quinoline. In addition to a phenyl substituent (3m), elusive moieties in previous Minisci alkylative protocols such as bromide (3o), ketone (3n), ester (3p) and amide (3q)[28, 30–33] were very well tolerated, giving rise to the alkylated quinolines with good yields. Additionally, we envisioned that 2-methylquinolines could lead to the desired 4-alkylated products as well (3r-t). Consequently, various N-heteroarenes were tested, such as phenanthridine (3u), benzothiazole (3v) and benzimidazole (3w), which were selectively alkylated at the most electrophilic position in good yields. Moreover, in order to extend the applicability of the method, other recognizable quinolines were also studied under these electrochemical conditions. Thus, late-stage alkylations of complex natural products decorated with various functionalities as quinoxyfen (fungicide, 3x), quinine (3y), cinchonidine (3z) and (S)-(+)-Camptothecin (antitumor activity, 3aa) provided the corresponding C2 or C4 alkylation products in efficient fashion. Notably, the model reaction of lepidine (1a) proceeded efficiently starting from 1 mmol (upscaling 10 times) and led to the desired alkylated quinoline derivative 3a in good yield (75%, see Supplementary Information).
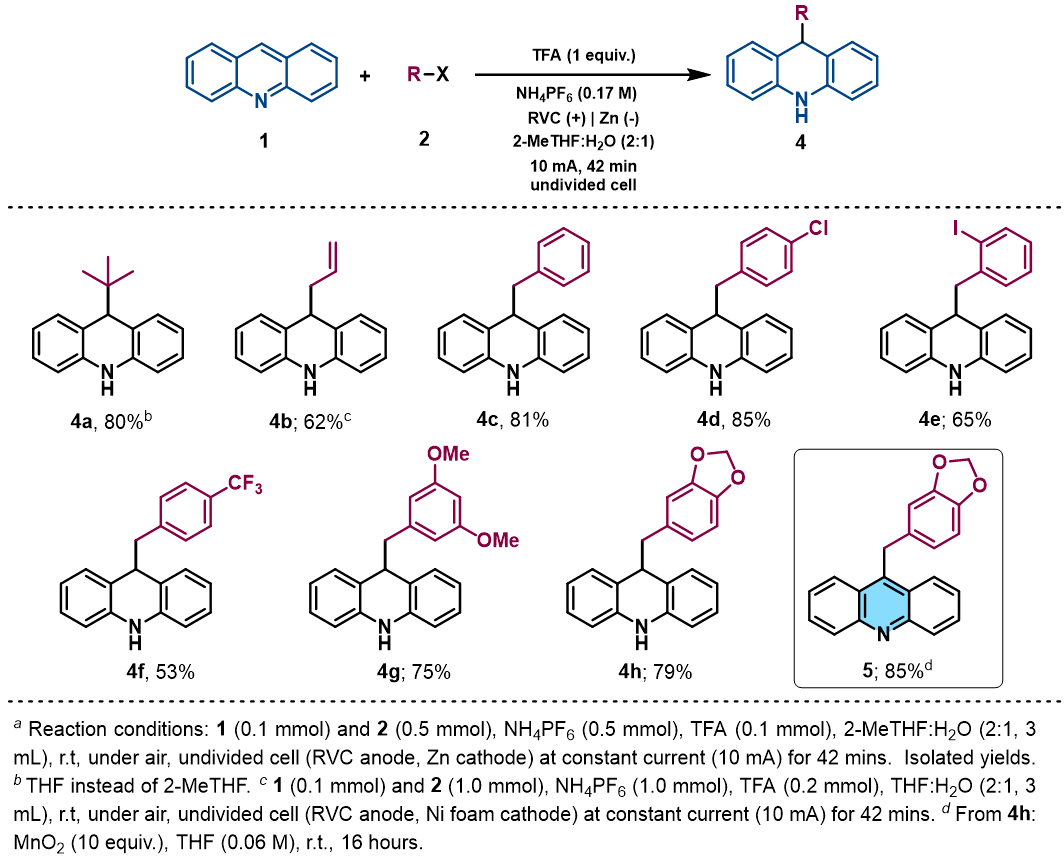
With the idea to expand the applicability of the method, we identified acridines as potential substrates for their selective and straightforward alkylation at C9 by using the presented electrochemical methodology. Acridine derivatives constitute a class of compounds with a broad spectrum of biological activity and are of great interest for the organic and medicinal chemistry field., Therefore, due to the lack of straightforward approaches to accomplish the direct functionalization of acridines in the literature, the synthetic modification of this prized heterocyclic core could be particularly appealing. Gratifyingly, under slightly modified reaction conditions (see Supplementary Information for details), acridine (1v) was found to be a suitable alkyl radical acceptor. Thus, the tert-butyl radical was added to the C-9 position of the acridine, giving rise to the corresponding dihydroacridine derivative in good yields (Table 3, 4a).
We next examined the allylation and benzylation of acridine, which would give access to products whose synthesis, to the best of our knowledge, has never been accomplished by a Minisci-type alkylation protocol. Therefore, allyl bromide was efficiently reduced under the electrochemical reaction conditions and led to the desired allylated product 4b in good yield. Different benzyl bromides with varying electronic nature (4c-h) were tolerated using 2-MeTHF (included in the “green” solvent list) as an environmentally friendly solvent. Notably, synthetically versatile halides such as chloride (4d) and even iodides (4e) were compatible under the electrochemical reaction setup, leading to the benzylated dihydroacridine derivatives in good yields. Furthermore, the system was easily rearomatized under mild oxidative conditions to give the corresponding C9-functionalized acridine (5) in excellent yield.
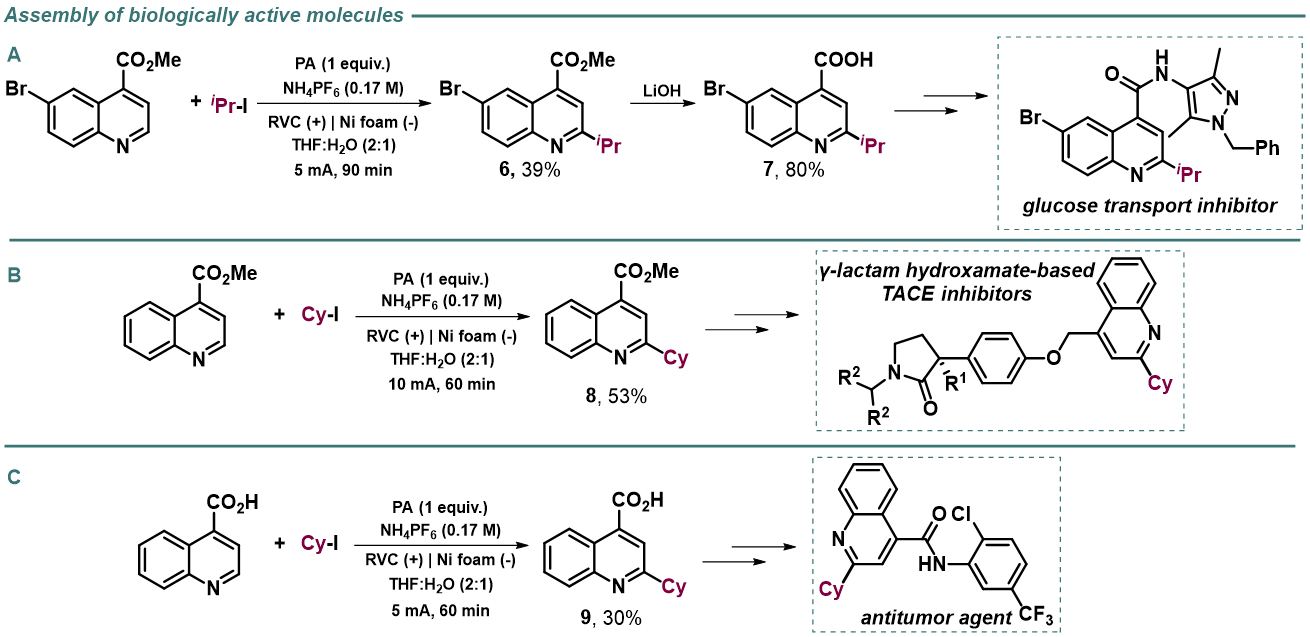
As shown before, interesting and versatile carboxylic acid derivatives allocated at the 4-position of the quinoline were remarkably compatible under the electrochemical reaction conditions (Table 2). Encouraged by these results and in attempt to expand the applicability of the method, we targeted the formal synthesis of various interesting drug molecules. Thus, the iso-propyl installation at the 2 position of ester-bearing quinoline 1h using our optimized electrochemical protocol led to the desired alkylated quinoline 6 with excellent selectivity. Simple hydrolysis of the ester moiety gave rise to carboxylic acid derivative 7, which has been used as a template for the construction of a more complex amide-substituted quinoline featured as a glucose transport inhibitor (Scheme 1A). In addition, alkylated product 8, achieved directly following the electrochemical procedure, gave direct access to an intermediate in the synthesis of a tumor necrosis factor-α-converting enzyme (TACE) inhibitor (Scheme 1B). To our delight, this electrochemical system could also provide the corresponding alkylated quinoline 9 in the presence of a free carboxylic acid in synthetically useful yield, which is itself an intermediate in the synthesis of an antitumor agent (Scheme 1C) or used as template for the construction of anti-tuberculosis agents.
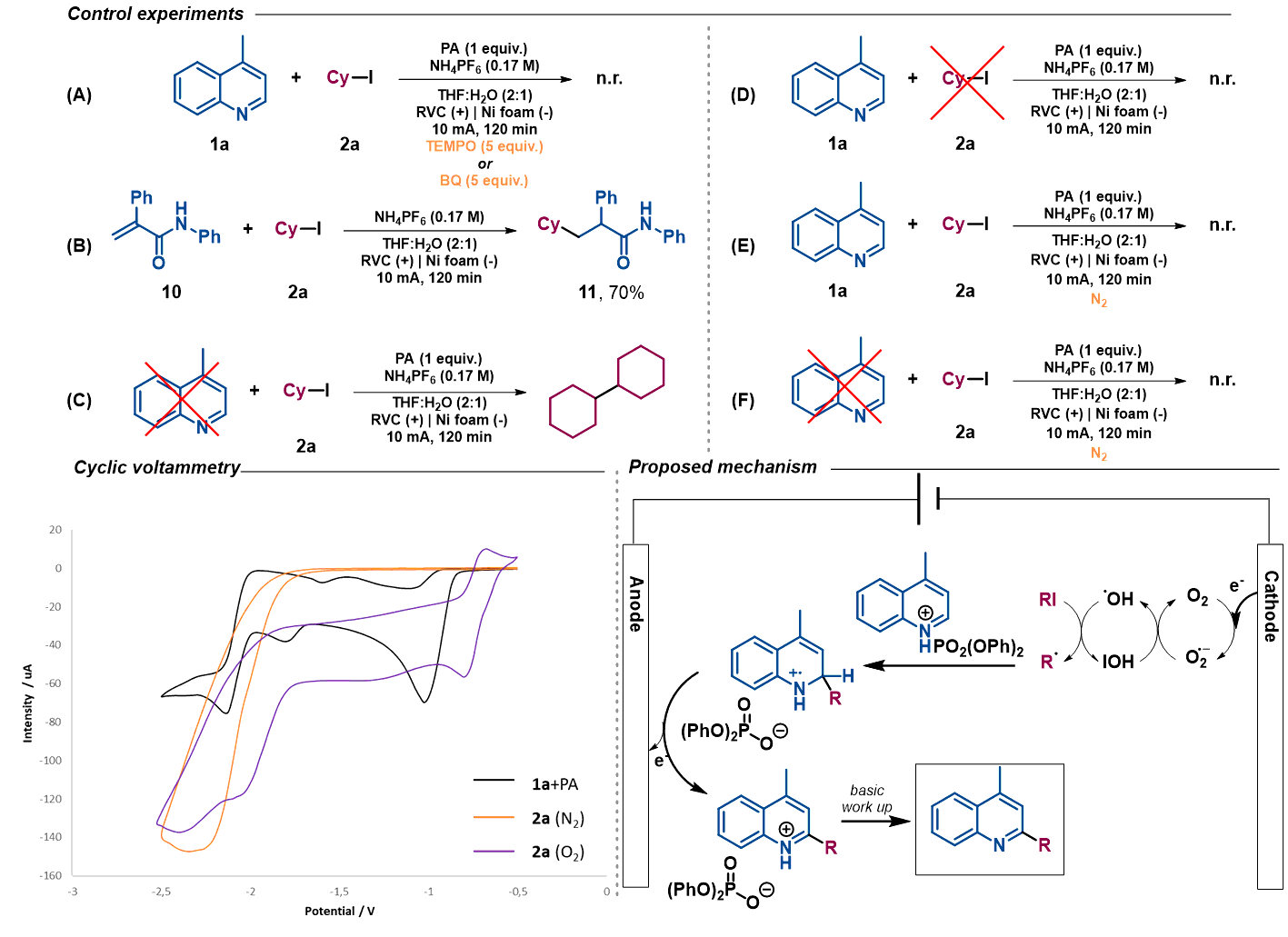
Mechanistic studies. We then performed a series of preliminary control experiments in order to gain an insight into the reaction mechanism. As shown in Scheme 2, in the presence of a radical scavenger as TEMPO (2,2,6,6-tetramethylpiperidinooxy), the reaction was inhibited as only starting materials were observed untouched (Scheme 2A). In addition, when an acrylamide derivative (10) was used as radical acceptor41,47 instead of lepidine, the corresponding Giese-type product was obtained in good yield (11) (Scheme 2B). These results strongly indicate that radical species are involved in the reaction pathway. When the reaction under the standard conditions was carried out in absence of lepidine (1a), the alkyl halide was completely consumed, and only the homocoupling product, resulted from the cyclohexyl radical dimerization, was observed (Scheme 2C). However, when performed in absence of 2a, lepidine activated with PA did not suffer any change or reduction process (Scheme 2D), which shows how the optimized electrochemical reaction conditions are unable to reach the required reduction potential (with and without PA, see S.I.) for such transformation (see cyclic voltammetry Scheme 2). Therefore, the PA is only enhancing the reactivity of the C=N bond.17 This may show a reductive pathway for the aliphatic halide, which might be generating carbon-centered radical species involved in the C-C bond forming event to yield the alkylated products. In addition, we tested the reaction under inert atmosphere conditions in order to evaluate if oxygen may be involved. In fact, under such conditions the reaction did not take place and only the starting materials were observed (Scheme 2E). Moreover, as expected based on the high reduction potential shown by halide 2a (see cyclic voltammetry Scheme 2), when a control experiment in absence of lepidine was carried out, no homocoupling product was observed (Scheme 2F). Therefore, oxygen should be involved in the generation of the initial reactive radical species. Moreover, as shown in Scheme 2A, in the presence of a superoxide scavenger as benzoquinone (BQ), the reaction was inhibited and the radical precursor was observed untouched.
Based on the above-mentioned findings and supported by the literature,47,40 we propose the mechanism shown on Scheme 2. We suggest that aerobic oxygen is responsible for the initiation of the process. Upon reduction, shown feasible with the lowest reduction potential of the reaction components (see cyclic voltammetry Scheme 2), the superoxide anion is formed and protonated to generate highly reactive peroxy radical species. These intermediates could be responsible for the generation of the carbon-centered radical via halogen atom abstraction of the alkyl halide. Following alkyl radical generation, addition to the activated (protonated) N-heteroarene would result in the formation of a new carbon-carbon bond. Finally, the putative radical intermediate would then undergo rearomatization to deliver the final Minisci-type adduct.
In conclusion, we have described a general and facile Minisci-type alkylation of N-heteroarenes under simple and straightforward electrochemical conditions using available alkyl halides as radical precursors. The reaction system has demonstrated its robustness and generality as primary, secondary and tertiary alkyl radical precursors and a variety of heterocycles have shown their compatibility within this electrochemical system. In addition, various heterocyclic-based natural products have been successfully integrated in the reaction scope. Moreover, as a consequence of the high functional group tolerance of the method, we have shown how the electrochemical Minisci-type alkylation methodology can be efficiently used to reach to biologically valuable compounds.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}